

本文作者:杉杉
导读
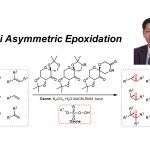
近日,斯坦福大学Barry M. Trost教授课题组在Angew. Chem. Int. Ed.上发表论文,报道了一种新型的Pd(0)催化区域和对映体选择性[3+2]螺环反应,从而合成相应的[5,5]螺环碳环和杂环化合物。值得注意的是,区域选择性可通过Pd-π-烯丙基中间体来控制。同时,该反应具有广泛的底物范围、出色的区域选择性和对映选择性。
Regiodivergent Synthesis of Spirocyclic Compounds through Pd-Catalyzed Regio- and Enantioselective [3+2] Spiroannulation
Barry M. Trost and Zhijun Zuo
Angew. Chem. Int. Ed. ASAP DOI:10.1002/anie.202016439
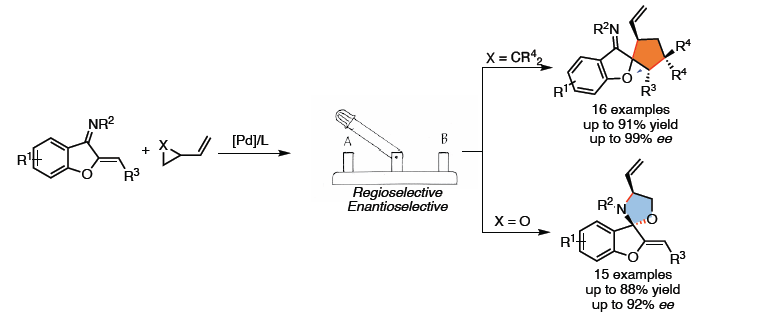
正文
作为常见的有机反应,α, β-不饱和酮或亚胺的亲核加成反应可分别进行1,2-加成和1,4-加成(Scheme 1a)。环加成反应,作为一种直接合成复杂化合物的通用策略(具有步骤经济性)。其中,由活性两性离子中间体和不饱和受体组成的1,3-偶极环加成(1,3-DCs),在合成具有价值的碳环和杂环中发挥重要的作用,但常规可用的1,3-偶极子类型有限。氮杂二烯1作为偶极环加成反应中有效的亲偶极子(dipolarophile),其结构上具有独特的顺式构型和苯并呋喃核上的亚胺单元。在过去几年中,氮杂二烯已作为各种不对称迈克尔加成反应及其串联反应中的四原子合成子,可获得多种含苯并呋喃的环状化合物。最近,Trost课题组和其他课题组[1]报道了Pd催化的氮杂二烯的不对称[4+3]环加成反应,从而构建四氢氮杂䓬化合物(tetrahydroazapines)(Scheme 1b, top)。2017年,Zhao课题组[2]通过Pd催化氮杂二烯与1,5-偶极子之间的[5+4]环加成反应,以优异的对映选择性合成苯并呋喃稠合的九元N-杂环化合物(Scheme 1b, middle)。随后,使用1,6-偶极子[3],也实现了十元N-杂环化合物的合成(Scheme 1b, bottom)。然而,对于Pd催化氮杂二烯的区域和对映选择性的[3+2]螺环化反应,尚未被报道。基于前期的研究成果[4],作者设想氮杂二烯是否也可作为对映选择性[3+2]螺环化反应中的有效合成子,从而合成具有价值的螺环化合物(作为天然产物和药物的核心骨架)(Scheme 1c)。同时,该反应也存在一定的挑战:(1)氮杂二烯底物易发生[4+n]环加成反应;(2)由于存在1,2-加成和1,4-加成的可能性,导致不可预测的立体选择性和区域选择性。
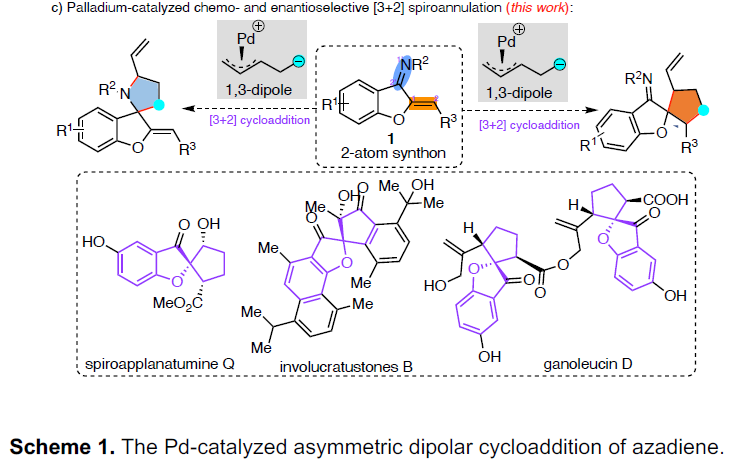
最近,也有文献报道[5]使用具有吸电子基团的乙烯基环丙烷(VCPs)衍生物作为1,3-偶极子前体。因此,作者以氮杂二烯1a和VCPs 2作为底物,对反应进行了研究(Scheme 2)。当在催化量的Pd2(dba)3•CHCl3和手性BINAP存在下,底物2a和1a进行了[3+2]螺环化反应以71%的收率得到螺环化合物,同时具有中等的非对映选择性和对映选择性。同时,具有不同空间和电子性质的底物2b–2d与体系不相容。值得注意的是,Meldrum酸衍生物2e,以优异的收率(83%收率)和对映选择性(97%ee)获得相应的产物3e。
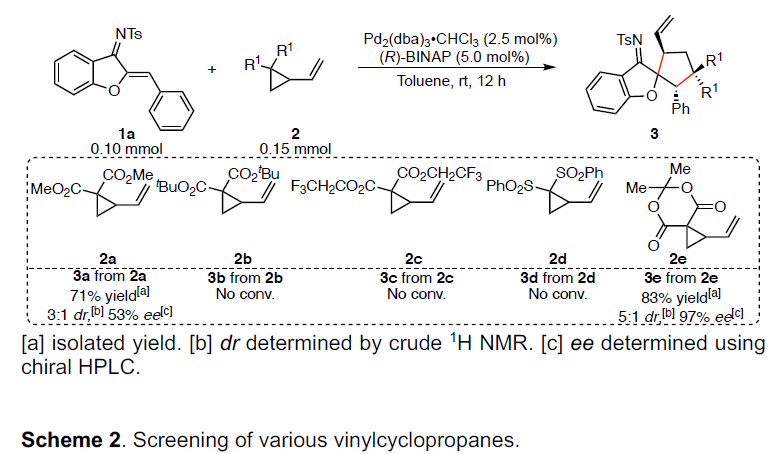
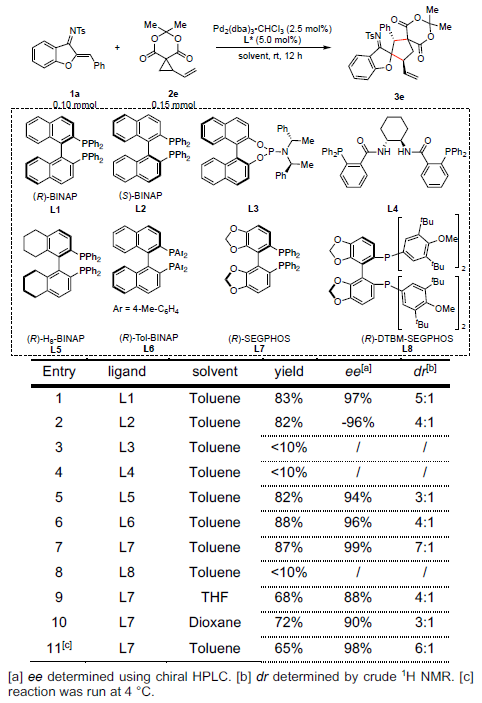
随后,作者以1a和2e作为模型底物,进行了相关[3+2]螺环化反应条件的筛选(Table 1)。反应结果表明,当以Pd2(dba)3•CHCl3为催化剂,L7为配体时,可在甲苯溶剂中室温反应,即可获得65%收率的目标产物3e(ee为98%和dr为6:1)。
![]()
在获得最佳反应条件后,作者开始对底物1和2进行了扩展(Scheme 3)。氮杂二烯底物1中芳基的对位和间位具有不同电子取代基底物,均能顺利进行反应,获得相应的产物3f–3m。值得注意的是,含有溴和氯等官能团也与体系兼容,为后期修饰提供了多种可能。同时,具有杂环、萘基底物,也以优异的收率和对映选择性获得了相应的产物3n–3p。并且,苯并呋喃核上的甲基对反应没有明显影响,以73%的收率和93%的ee得到相应的产物3q。此外,其他类型的VCPs,如双氰基型(2f),1,3-茚满二酮型(2g)和N, N-二甲基巴比妥酸型(2h)均可平稳地进行转化,从而以高收率和对映选择性获得螺环产物3r–3t。

随后,作者将底物2改为环氧乙烯基4a,再对氮杂二烯底物1的范围进行了扩展(Scheme 4)。通过再次筛选后发现,当使用(R)-BINAP作为配体时,可获得最佳反应结果。反应结果表明,芳基上的取代不受电子效应和定位效应的影响,均可获得较高收率和优异对映选择性的产物5a–5k。同时,含杂芳基的氮杂二烯也与体系相容(5l–5m)。此外,苯并呋喃核上的甲基和甲氧基对反应没有明显影响,均可获得相应的产物5n和5o。

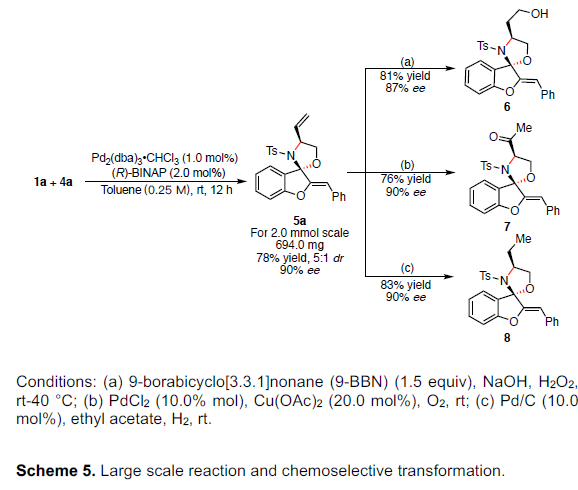
紧接着,作者对反应的实用性进行了研究(Scheme 5)。首先,将反应放大至2 mmol时,可将催化剂的负载量降至1.0 mol%,5a的收率和ee均保持不变(Scheme 5, left)。其次,5a可进行相关的衍生化。如5a的末端烯烃可被氢化/氧化以高收率得到相应的醇6,尽管ee略有降低(Scheme 5a)。5a经Wacker氧化以76%的收率获得甲基酮7,对映选择性不受影响(Scheme 5b)。5a的末端烯烃经Pd/C还原后,以83%的收率获得产物8,ee没有损失(Scheme 5c)。
根据相关的实验结果和文献[6],作者提出了这种高度区域选择性[3+2]螺环化的合理过渡态(Scheme 6)。Pd(0)物种的配位和氧化加成到乙烯基环丙烷或乙烯基环氧化物中,生成两性离子的Pd-π-烯丙基中间体。当X是双取代的碳时,Pd-π-烯丙基中间体更易将碳亲核试剂进行1,4-加成至氮杂二烯,以使电荷离域化最大化(后期过渡态)。随后,中间体II中的空间相互作用导致酮亚胺阴离子C进攻π-烯丙基配合物生成产物3。当X是氧时,Pd-π-烯丙基中间体的氧亲核试剂更易进攻电子空间不足,位阻更大的氮杂二烯酮亚基(早期过渡态),形成中间体II’,然后通过环化得到的氮阴离子从而获得产物5。

总结
斯坦福大学Barry M. Trost教授课题组报道了一种新型的Pd(0)催化区域发散性和对映体选择性[3+2]螺环反应,从而合成相应的[5,5]螺环碳环和杂环化合物。值得注意的是,区域选择性可通过Pd-π-烯丙基中间体来控制。同时,该反应具有广泛的底物范围、出色的区域选择性和对映选择性,并且通过对产物的后期修饰进一步证明了反应的实用性。














No comments yet.