
作者:石油醚
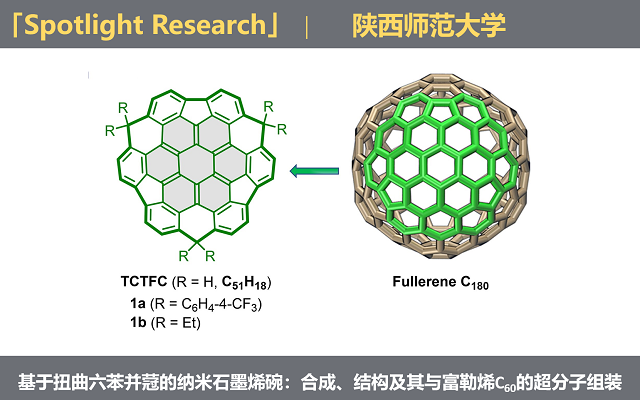
2026年5月1日,Science Advances在线发表了来自陕西师范大学魏俊发教授团队的题为“Nanographenic bowls based on contorted hexabenzocoronene: Synthesis, structure, and supramolecular assembly with fullerene C60”的研究论文。该工作首次在溶液中成功合成了基于扭曲六渺位六苯并蒄 (hexa-cata-hexabenzocoronene, c-HBC) 骨架的全边缘闭合的纳米石墨烯碗。研究团队开发了一种“支化臂环化”(Branching Arms Cyclization, BAC) 合成策略,攻克了c-HBC基巴基碗长期存在的溶液相合成难题,为拓展该类纳米石墨烯碗家族提供了强有力的新范式。
本期热点研究,我们邀请到了本文第一作者,来自陕西师范大学的博士后孙一洵为我们分享。
巴基碗 (buckybowls) 或称π-碗,其纳米级同系物也被称为纳米石墨烯碗,因优美的曲面π共轭结构和广阔的应用前景,一直是合成化学家们追逐的“明星分子”。然而,当碗的尺寸变大、π表面大幅扩展时,芳香体系为了维持平面化而产生的巨大环张力以及溶解度问题使得合成变得异常困难。目前已知的具有三层碳骨架的大尺寸纳米石墨烯碗的数量极其稀少,主要为基于心环烯骨架的碳纳米锥和基于平面六迫位六苯并蒄 (hexa-peri-hexabenzocoronene, p-HBC) 骨架的超级花烯衍生物。相比之下,p-HBC的“孪生姊妹”——双凹面的c-HBC骨架,虽然天然具有形成碗状结构的潜力,但由于合成上更加棘手、湾区空间位阻极大,此前,研究人员仅能在金属Ru表面通过表面合成法捕捉到相关踪迹 (图 1),或借助溶液化学合成带有明显缺口的不完整结构。因此,如何精准构筑基于c-HBC骨架且具有完整封闭边缘拓扑的纳米石墨烯碗,始终是该领域未解的重大挑战。我们课题组多年深耕于新型纳米石墨烯碗的设计合成,对这个悬而未决的难题抱有浓厚兴趣。
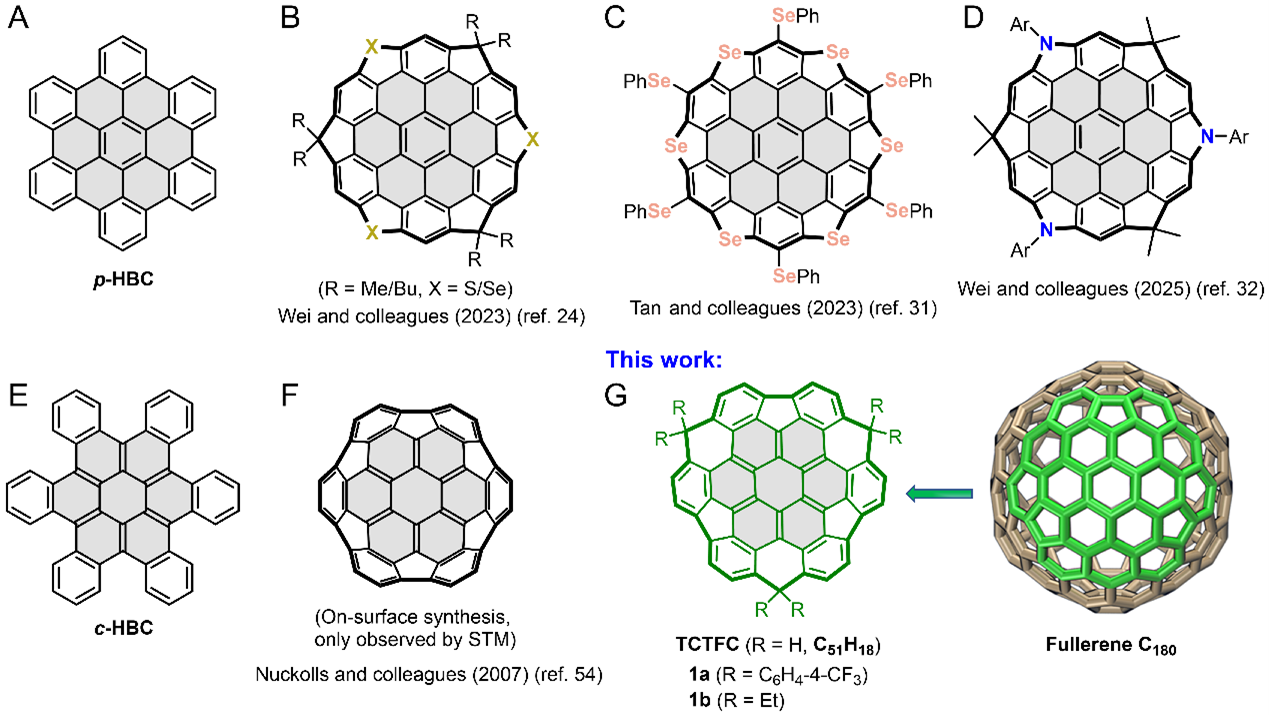
图1. 代表性HBC基巴基碗的化学结构与本工作目标分子TCTFC。
如果按照常规思路,先构建一个双凹面的c-HBC骨架,再想办法把它“掰弯”,需要直面c-HBC骨架湾区巨大的空间位阻和环张力。受之前合成三硫族杂超级花烯衍生物的启发 (Nat. Commun. 2023, 14, 3446),我们意识到sp3桥碳可作为“分子锚”来锁定正曲率。为此,我们设计了BAC策略,核心思路是在合成的最早期就把三个sp3桥碳单元预装到蒽酮类构筑模块中。这些模块如同携带了全部施工信息的“臂膀”——预置的硼酸酯、羰基和氯原子,依次完成中心核偶联、蒄核构筑及湾区五元环闭合。该策略巧妙规避了直接向c-HBC骨架引入桥碳单元的巨大位阻,成功获得了目标分子——三碳环化三芴并蒄 (tricarbon-annulated trifluorenocoronene, TCTFC) 衍生物1a和1b (图2)。
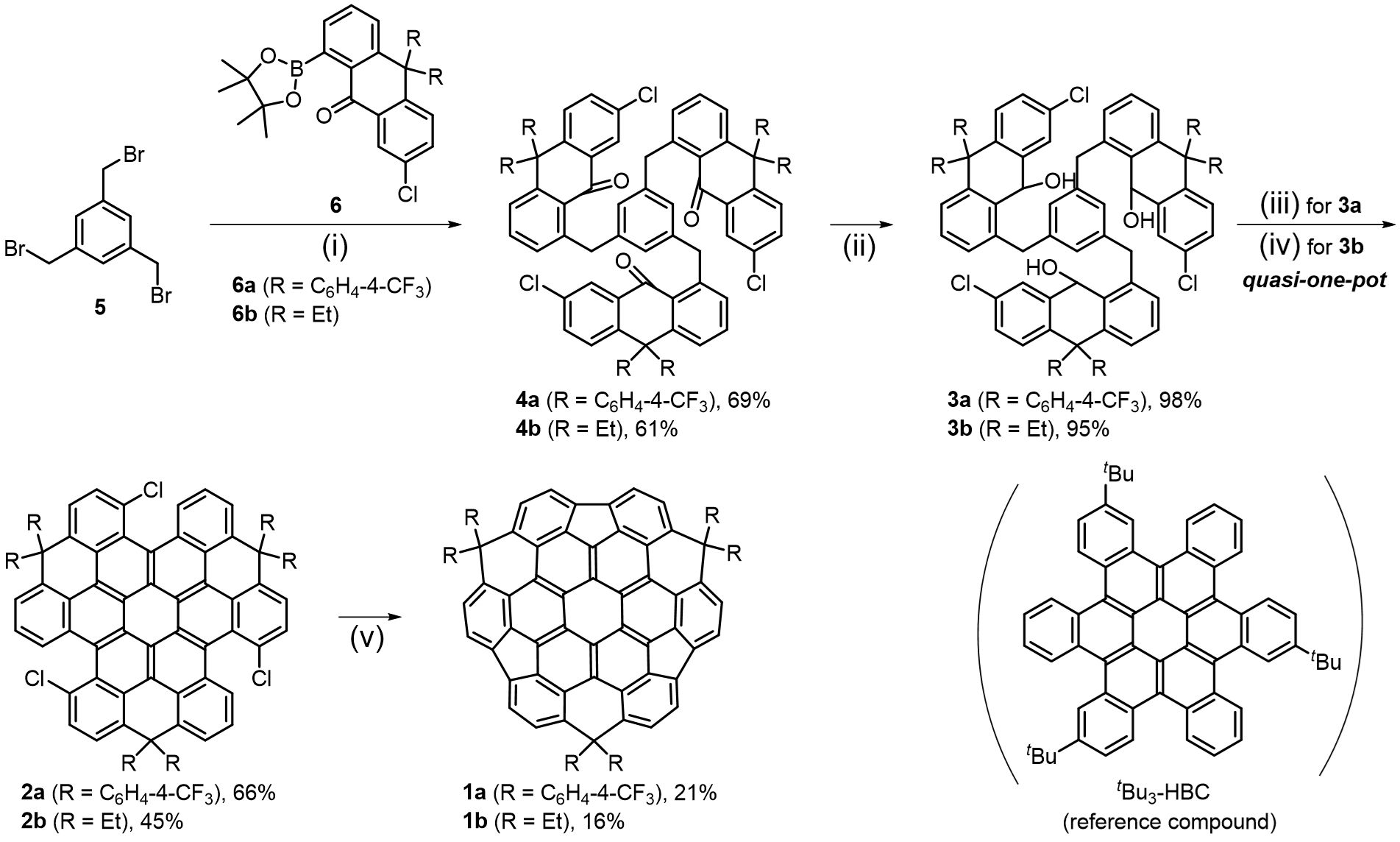
图2. 纳米石墨烯碗1a和1b的合成路线。试剂和条件:(i) Pd(PPh3)4, Cs2CO3, toluene/H2O, 80°C, overnight; (ii) NaBH4, THF/MeOH, r.t., 2 h; (iii) For 3a:1) BF3·Et2O, DCM, r.t., 2 h; 2) DDQ, DCE, 80 °C, 30 min; 3) TfOH, 0 °C, 15 min; (iv) For 3b: 1) BBr3, DCM, r.t., overnight; 2) DDQ, DCE, 80 °C, 30 min; 3) TfOH, 0 °C, 15 min; (v) PdCl2(PCy3)2, DBU, DMAc, 145 °C, overnight.
X射线单晶衍射分析清晰揭示了1a和1b的准C3对称的深碗构型 (图3),碗深达到2.3 Å,直径超过1 nm。π轨道轴向量角 (POAV) 分析显示与中心轮毂环相连的五元环碳原子具有显著的锥化角度 (6.8° ‒ 7.2°)。HOMA与NICS计算表明碗分子呈现“外强内弱”的芳香性分布特征:外围六元环保持强芳香性 (HOMA = 0.85;NICS(1)zz = -18.29 ppm);中间六元环的芳香性减弱 (HOMA = 0.67 ‒ 0.68;NICS(1)zz = -12.27 ppm);而中心轮毂环则表现为反芳香性 (HOMA= 0.06 ‒ 0.36;NICS(1)zz = 11.30 ppm)。ACID与GIMIC计算进一步揭示了内外对旋的拓扑环电流模式。与c-HBC母体相比,TCTFC的碗状弯曲结构削弱了中心与边缘之间的电子耦合,并减弱边缘的π电子离域。
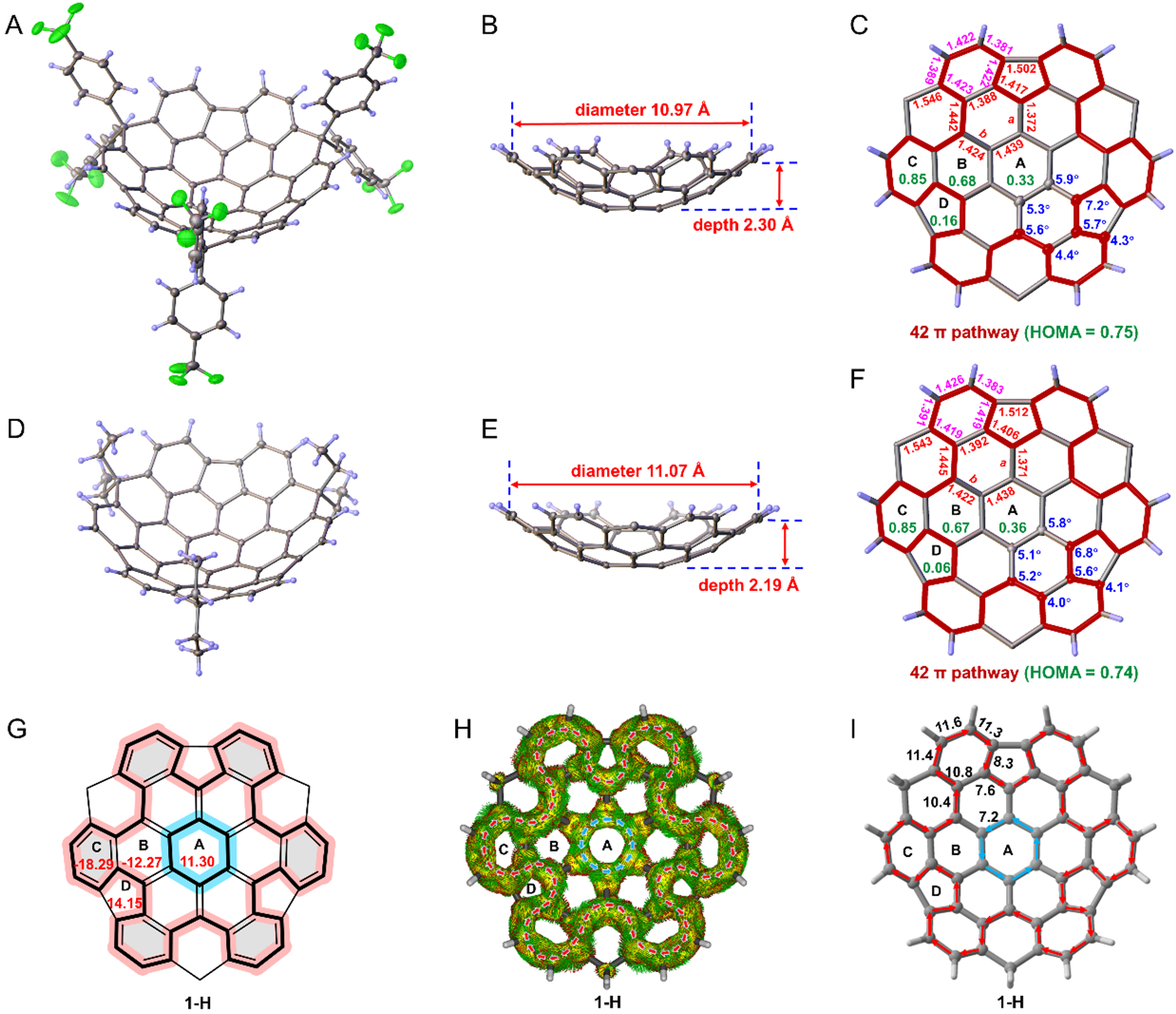
图3. 1a和1b的X射线单晶结构、POAV分析及芳香性评估。
我们通过变温1H NMR实验结合DFT计算研究了碗分子的构象动力学行为 (图4)。1a中凹面芳基的核磁信号在413 K时发生融合,通过Eyring方程计算得到实验旋转能垒为28.8 kcal/mol,与DFT计算结果 (22.2 kcal/mol) 定性吻合。两种碗分子均表现出高的碗-碗翻转能垒:模型化合物1-H的理论翻转能垒 (ΔGin = 56.4 kcal/mol) 远超经典碗烯 (9.1 kcal/mol) 和花烯 (16.3 kcal/mol),这归因于其显著更深的碗深 (2.23 Å) 所导致的巨大应变积累。
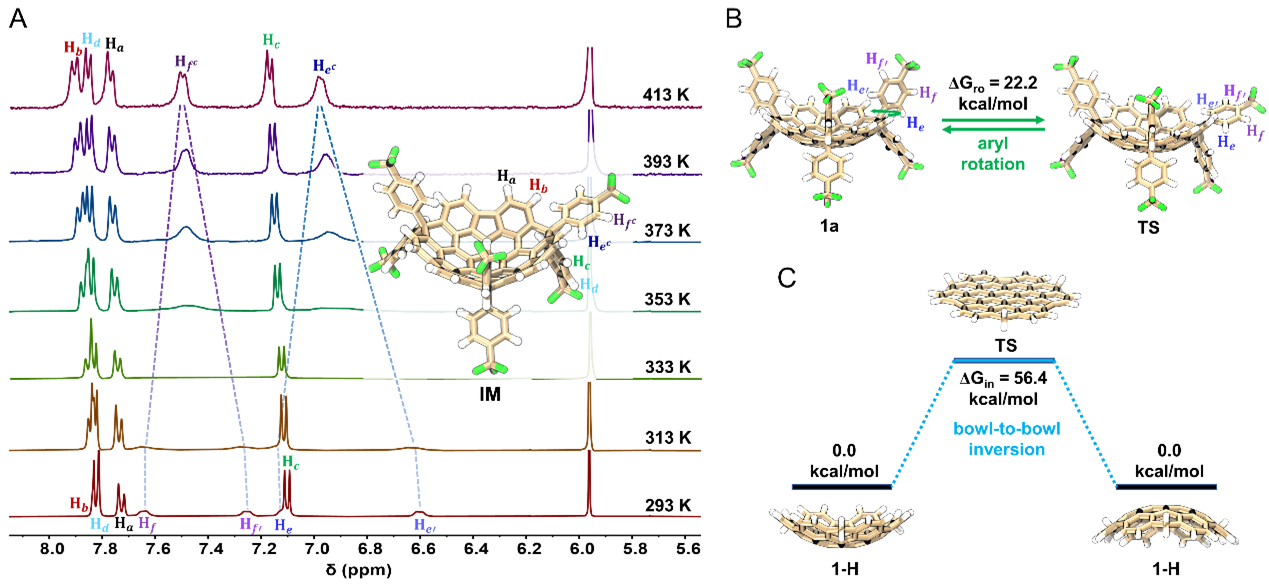
图4. 1a的变温核磁实验及理论计算的阻转和碗-碗翻转能垒。
光物理研究表明,与参比化合物tBu3-HBC相比,碗分子1a和1b的吸收与发射光谱均发生红移且强度减弱,光学能隙也相应收窄 (图5)。TD-DFT计算表明,碗状弯曲改变了前线轨道的跃迁模式,使允许跃迁的振子强度大幅降低。在THF/水混合溶剂中,随着水含量的增加,两种碗分子均表现出聚集诱导荧光增强 (aggregation-induced emission enhancement, AIEE) 效应,其固态荧光量子产率高于溶液态。这一现象可归因于聚集态下分子运动受限,从而抑制了非辐射衰变通道。电化学测试进一步显示,碗分子1a的循环伏安曲线中呈现出两个可逆的还原峰,且还原半波电位 (-1.36 V) 相比于tBu3-HBC (-1.83 V) 大幅正移,表明LUMO能级显著降低。DFT计算揭示了背后的电子机制:碗分子1-H的LUMO实际上源自c-HBC的LUMO+2轨道,弯曲结构赋予其新的空间重叠,从而增强了电子亲和势。
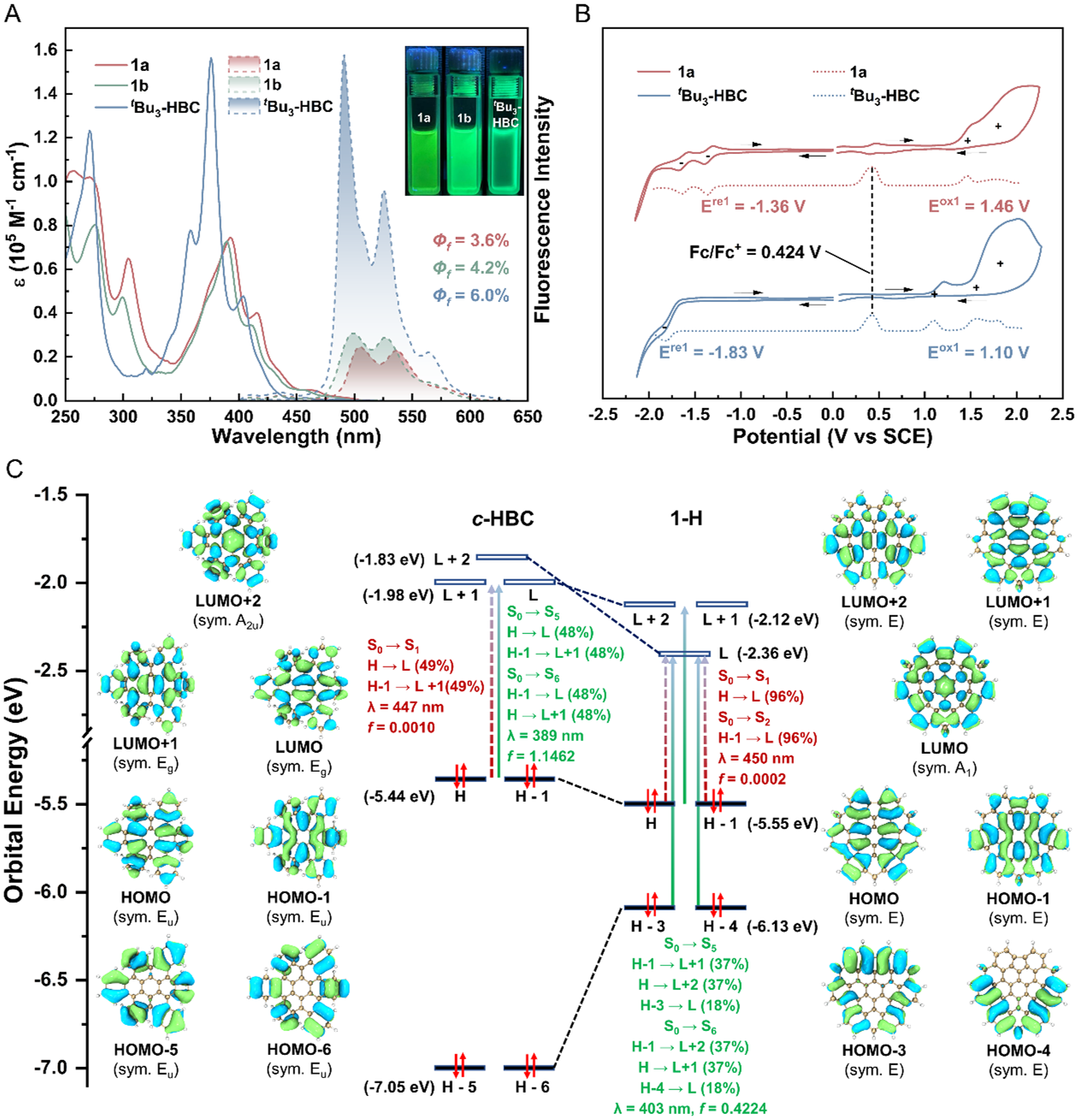
图5. 1a和1b的光物理与电化学表征及TD-DFT计算。
得益于深碗空腔与C60凸面的高度互补,1a和1b均表现出对C60较强的包结亲和力。1H NMR滴定实验显示两种碗分子与C60均呈现核磁时间尺度上的慢交换络合过程。在逐步滴加C60的过程中,1a较1b更早达到滴定终点,这直接预示了前者更强的络合倾向 (图6)。MALDI-TOF质谱确认了1:1络合物的形成,而且DOSY实验进一步证实络合后扩散系数一致性降低,表明溶液中形成了均一的主客体组装体。UV-Vis滴定实验测得1a的结合常数为 (2.31 ± 0.01) × 103 M-1,1b为(1.59 ± 0.002) × 103 M-1。该数值远超经典碗烯和花烯 (后者在溶液中与C60几乎不发生络合),在目前已报道的纯碳氢巴基碗受体中位居前列。值得注意的是,荧光滴定虽也观察到络合引起的猝灭现象,但受动态碰撞猝灭和内滤效应的共同影响,所得表观结合常数偏高,因此UV-Vis数据被视为更可靠的定量依据。1a表现出更强亲和力的原因在于其较深的空腔提供了更优的凸凹互补,凹面芳基在空间取向上更有利于与C60表面形成C–H⋯π接触,从而协同增大了有效接触面积。这一结论得到了IGMH等值面图与相互作用能计算的一致支持:1a@C60的接触面积更大 (221.63 vs. 217.91 Å2),总相互作用能也更负 (−45.7 vs. −41.9 kcal/mol)。SobEDAw能量分解进一步揭示,两体系中静电作用基本相当,尽管1a@C60的交换排斥略大,但被显著增强的色散作用和更有利的轨道相互作用完全补偿。
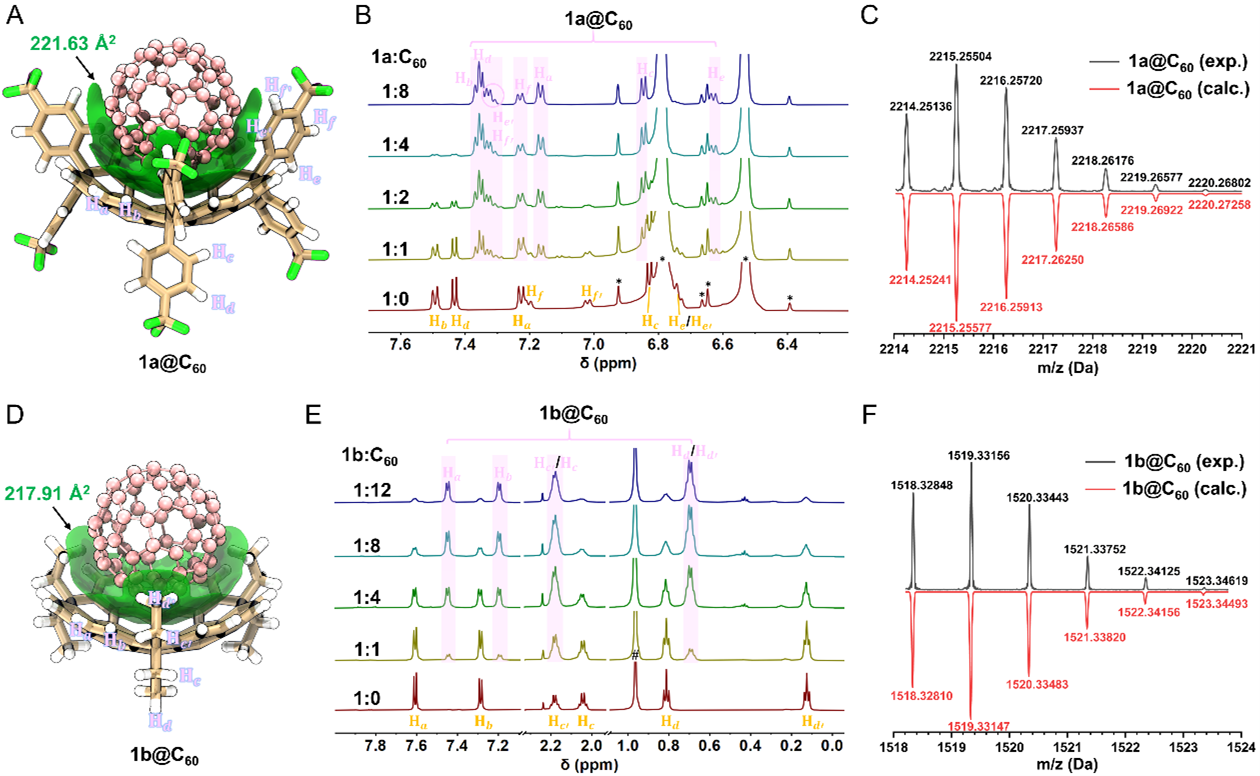
图6. 1a和1b与C60的主客体相互作用。
在固态共晶中,两种碗分子展现出截然不同的组装模式:1a与C60形成连续交替的柱状超分子堆积结构 (首次在碗-富勒烯体系中观测到);而1b则与C60组装成2:1的“三明治”夹心结构,并进一步扩展为三维蜂窝网络 (图7)。这种由外围取代基微妙调控的组装多样性,为超分子可控组装提供了经典范例。
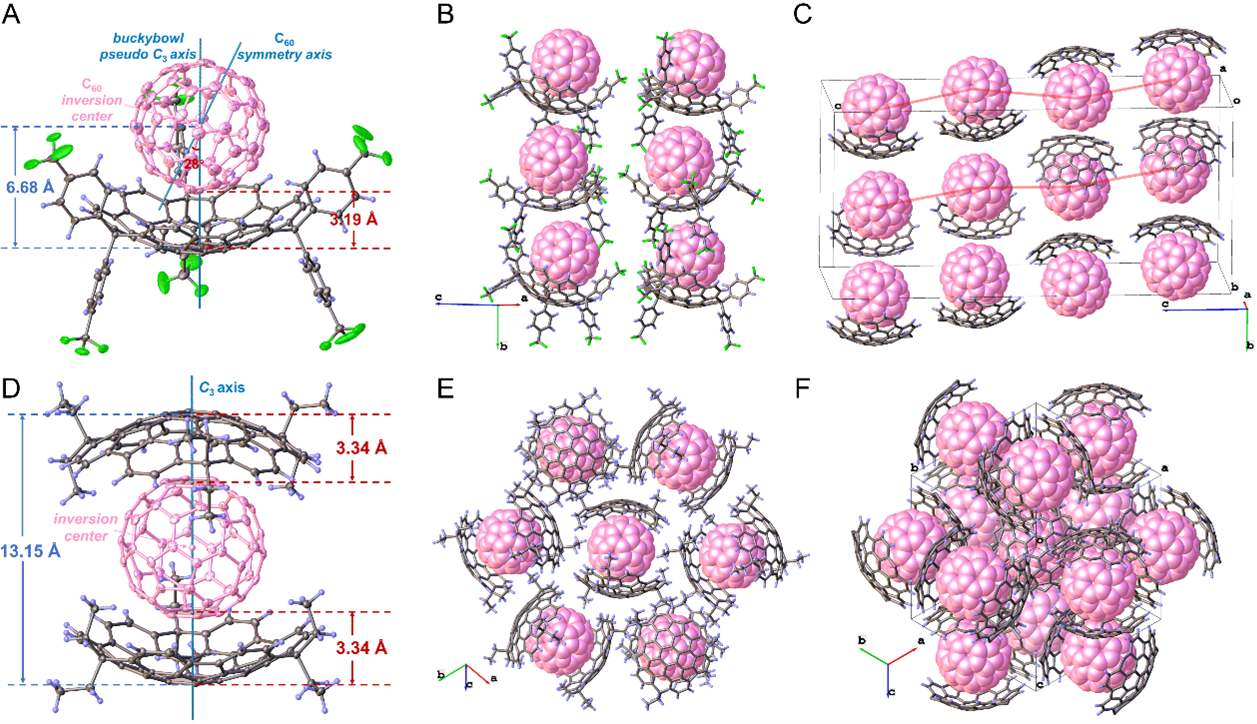
图7. 1a和1b与C60的固态组装结构。
综上,本研究发展了一种BAC策略,成功构建了一类基于c-HBC骨架的新型C3对称、以蒄为底的纳米石墨烯碗。这一溶液相合成方法攻克了c-HBC骨架全边缘闭环巴基碗长期悬而未决的合成难题。所得TCTFC 衍生物1a和1b具有显著的碗深与直径。与c-HBC类似物相比,它们展现出收窄的能隙、增强的电子亲和力以及红移的光学特征。两种碗分子在溶液中与C60形成1:1主客体络合物,其包结亲和力与固态堆积模式可被外围取代基精细调控。BAC策略的成功实施,为设计三杂原子环化的纳米碗、扩展π表面的结构及其他具有定制电子与超分子性质的弯曲纳米碳材料打开了大门。
Q1:有关本次研究的时候遇到过怎样的困难呢?又是怎样克服的呢?
最大的困难毫无疑问是合成。1a从三醇中间体3a出发,用BF3·Et2O就能顺利完成Friedel-Crafts环化,三步总收率66%。但1b却完全不同。同样条件下3b几乎得不到目标产物,反而生成了一大堆复杂的副产物。经过反复尝试,我们终于发现用BBr3替代BF3·Et2O后,反应变得干净了许多,最终以45%的收率拿到了关键前体2b。这个经历让我深刻体会到,在合成化学中,底物的微小差异就可能彻底改变反应路径,必须根据实际情况灵活调整策略,而不是简单地照搬经验。
Q2:这项工作为c-HBC基巨型巴基碗的合成打开了大门。您认为下一步可以朝哪些方向拓展?
BAC策略的建立意味着我们有了一个通用平台。接下来,我认为有几个方向非常值得探索:第一,杂原子掺杂。我们之前已经在p-HBC体系中做了硫、硒、氮、硅的掺杂工作,证明杂原子可以显著调控碗分子的光电性质和组装行为。将类似的思路移植到c-HBC体系中,应该会带来更多有趣的发现。第二,进一步扩展π表面。目前我们做的是三层环绕结构,如果能把环绕层数扩展到四层,碗的尺寸会更大,曲率更接近富勒烯,对更大尺寸富勒烯 (比如C70、C84) 的识别能力可能会更强。第三,手性版本的构筑。c-HBC骨架本身具有手性潜力,如果能实现碗手性拆分,获得光学纯的纳米石墨烯碗,在手性发光材料方面将大有可为。
Q3:最后,有什么想对各位读者说的吗?
愿每一位深耕化学的追梦人,都能在浩瀚化学空间里,探索结构之美、解锁未知奥秘,坚守初心、潜心求索,在一次次沉淀与突破中,合成属于自己的璀璨征途。
作者教育背景简介
教育背景
2011-2015 广西师范大学化学与药学学院独秀实验班学士 (导师:潘英明 教授;潘成学 教授)
2016-2023 陕西师范大学化学化工学院博士 (硕博连读) (导师:魏俊发 教授)
2024-至今 陕西师范大学化学化工学院博士后 (合作导师:魏俊发 教授)
相关介绍
孙一洵,出生并成长于黑龙江抚远。2011-2015在广西师范大学化学与药学学院独秀实验班获得学士学位。2023年博士毕业于陕西师范大学,2024年进入陕西师范大学化学化工学院从事博士后研究,合作导师为魏俊发教授。主要研究方向为蒄基纳米石墨烯的合成及性质性能研究。目前以第一作者身份 (含共同第一作者) 在Science Advances、Nature Communications、Chem、Organic Letters、Advanced Synthesis & Catalysis期刊发表文章6篇,主持国家资助博士后研究人员计划 (C档)、中国博士后科学基金面上项目、陕西省博士后科研项目三等资助等。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/

















No comments yet.