
作者:杉杉
导读:
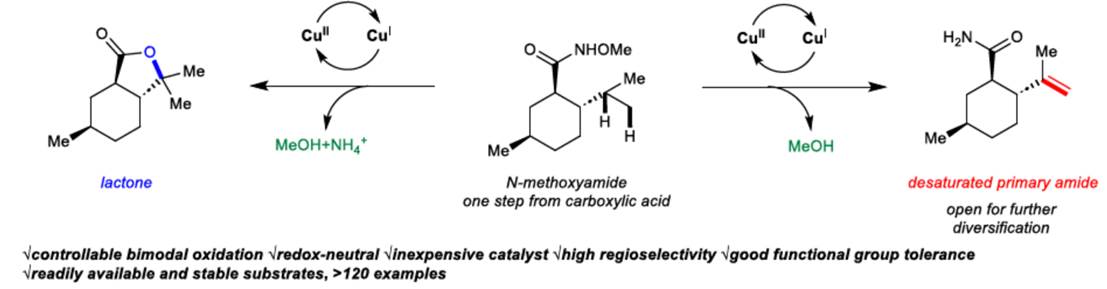
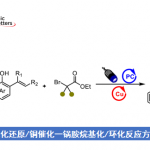
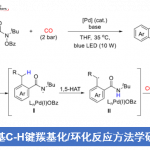
近日,美国Scripps研究所的余金权课题组在Nature中发表论文,报道一种全新的Cu(I)-催化N-甲氧基酰胺中γ-C(sp3)−H键的脱氢与内酯化反应方法学,进而成功完成一系列γ,δ-不饱和一级酰胺与γ-内酯分子的构建。其中,使用酰胺化合物作为自由基前体与内部氧化剂。
Copper-catalyzed dehydrogenation or lactonization of C(sp3)−H bonds
S. Zhou, Z. Zhang, J. Yu, Nature 2024, ASAP. doi:10.1038/s41586-024-07341-z.
正文:
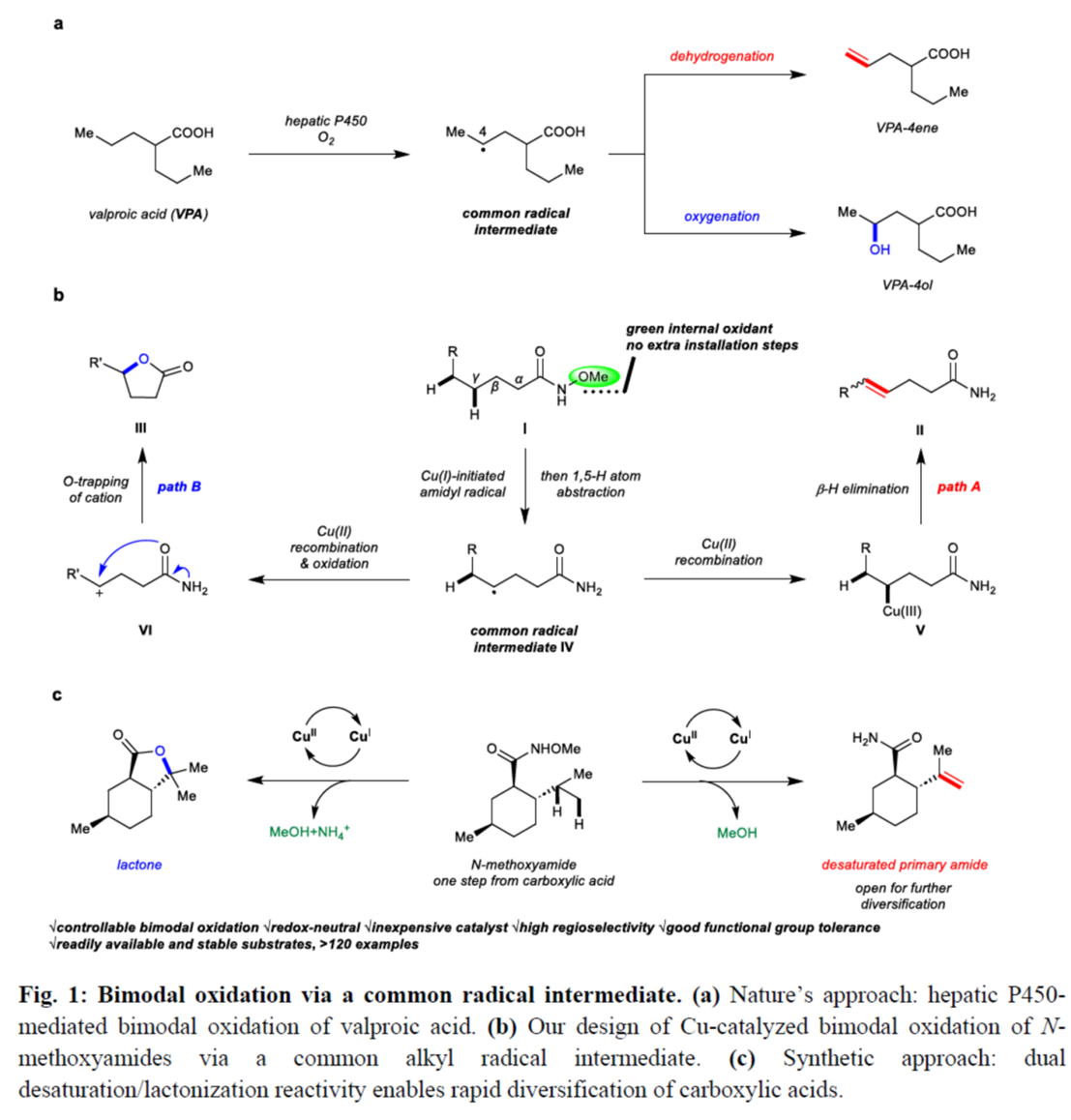
惰性的C−H键的直接氧化是构建C−O键的一种极具吸引力的策略。并且,通过C−H脱氢策略实现脂肪族链的去饱和反应[1],也同样具有价值。目前,化学家们利用hepatic cytochrome P450已实现了酶催化不同类型的反应方法学[2] (Fig. 1a)。同时,诸多研究团队已经成功设计出多种基于氢原子攫取(abstraction)策略实现了仿生脱氢与氧化反应方法学[3]。然而,此类反应常需使用外源氧化剂,并存在底物范围有限以及副反应多等弊端。受到N-甲氧基酰胺参与C−H活化反应方法学[4]、Cu(I)-介导活性肟酯生成亚胺自由基以及随后与tethered olefins进行环化反应方法学(Fig. 1b) [5]相关研究报道的启发,这里,美国Scripps研究所的余金权课题组报道一种全新的Cu(I)-催化N-甲氧基酰胺中γ-C(sp3)−H键的脱氢与内酯化反应方法学,进而成功完成一系列γ,δ-不饱和一级酰胺与γ-内酯分子的构建 (Fig. 1c)。
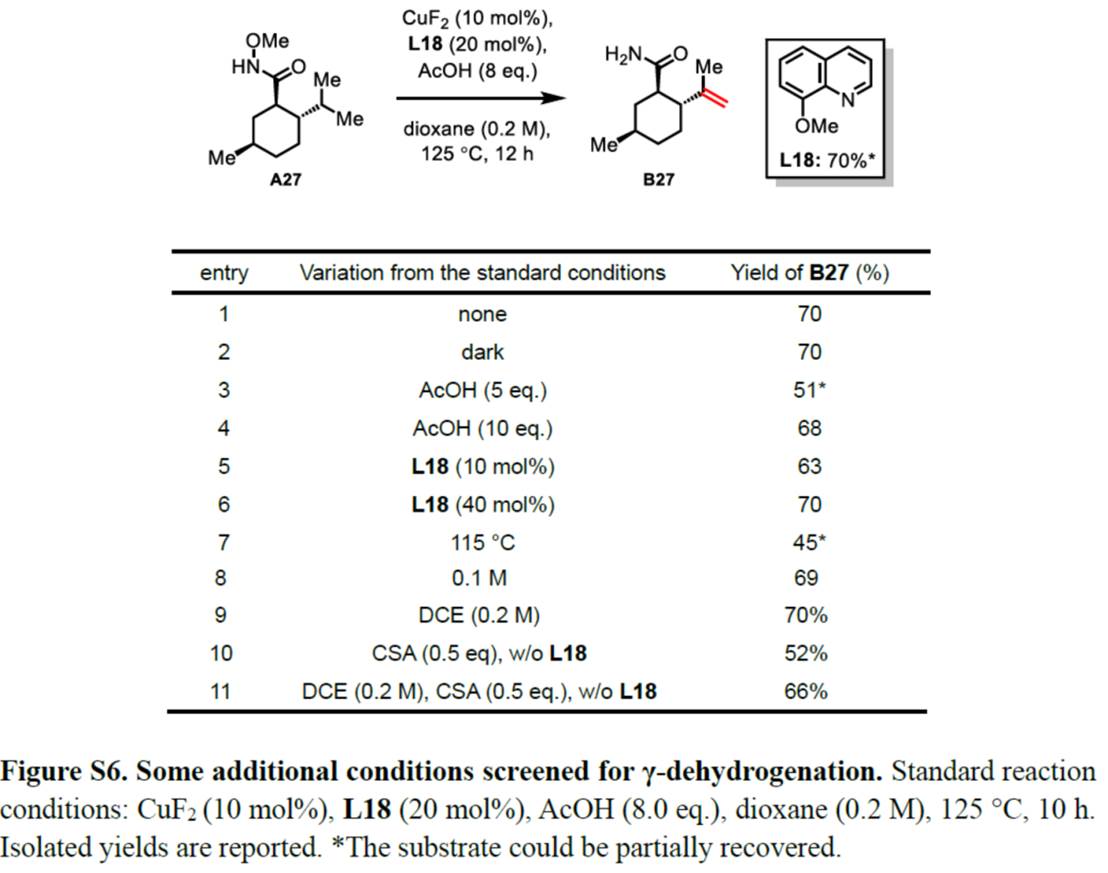
首先,作者采用N-甲氧基酰胺衍生物A27作为模型底物,进行相关脱氢反应条件的优化筛选 (Figure S6)。进而确定最佳的反应条件为:采用CuF2作为催化剂,8-甲氧基喹啉 (L18)作为配体,AcOH作为添加剂,在DCE或1,4-dioxane溶剂中反应,反应温度为125 oC,最终获得70%收率的脱氢产物B27。
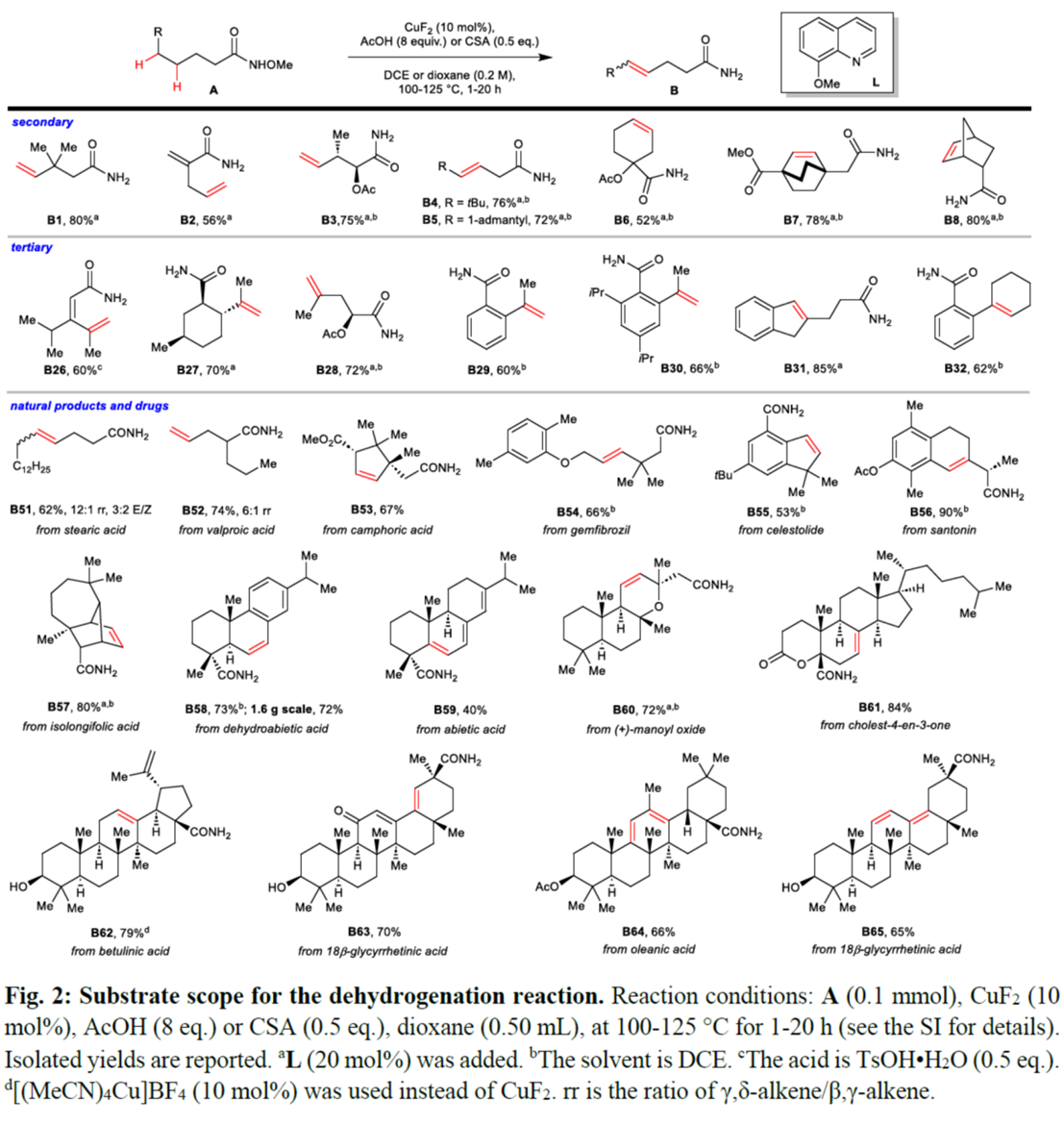
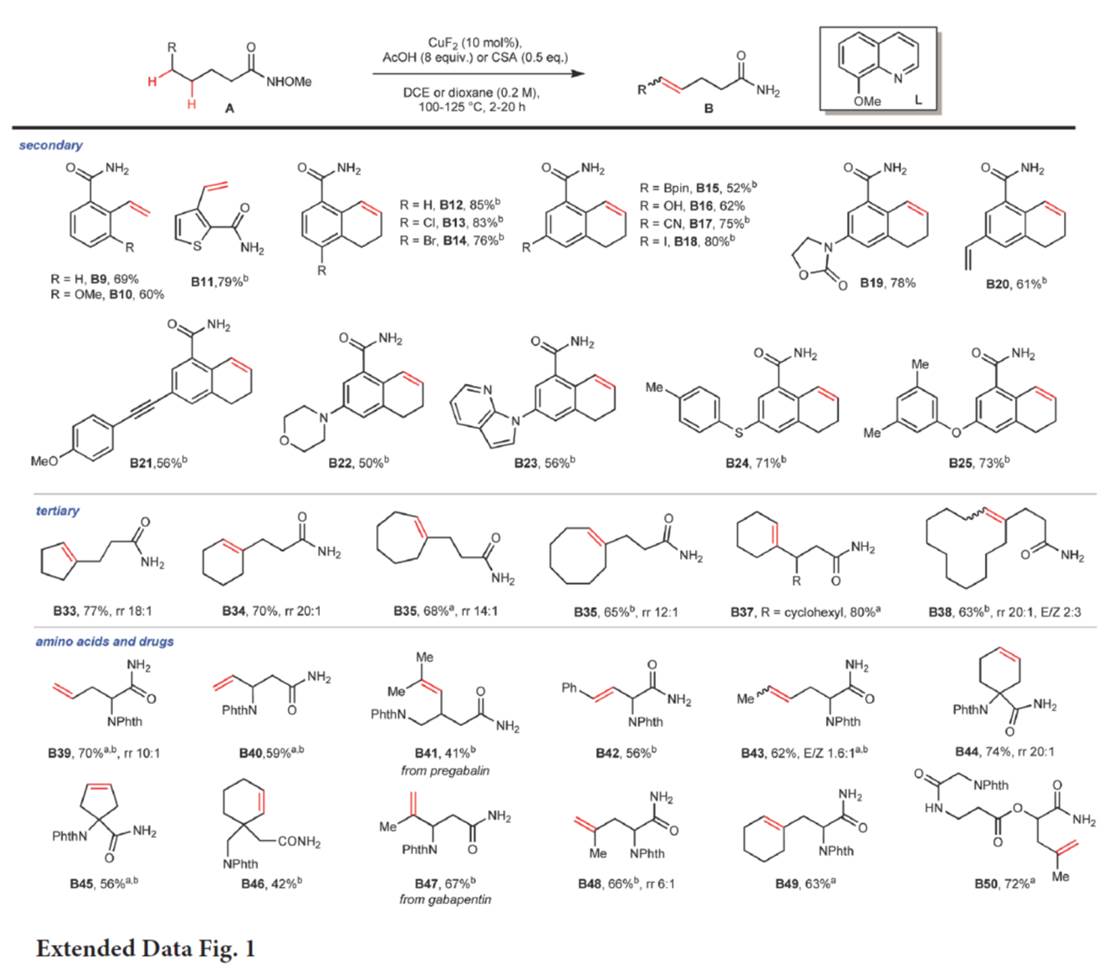
在上述的最佳反应条件下,作者对脱氢反应的底物 (Fig. 2与Extended Data Fig. 1)的应用范围进行深入研究。

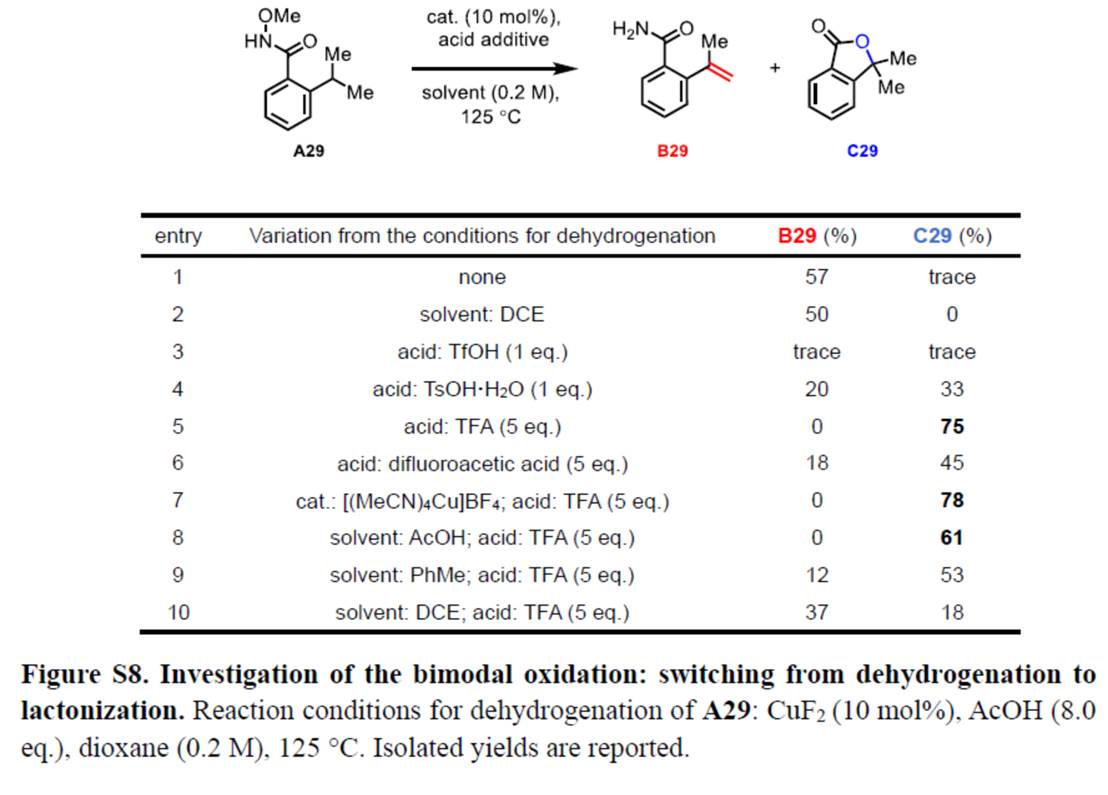
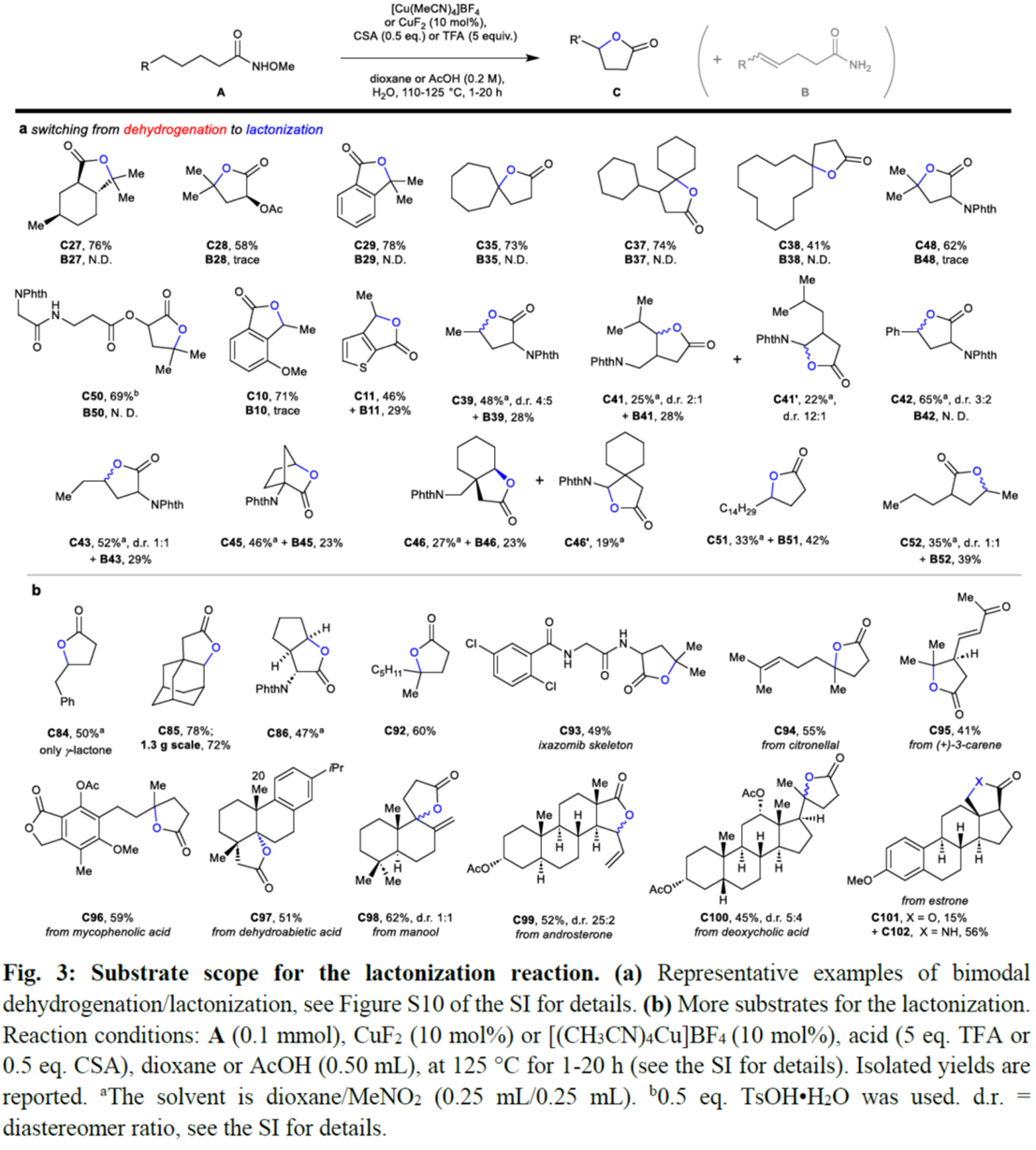
其次,作者采用N-甲氧基酰胺衍生物A29作为模型底物,进行相关内酯化反应条件的优化筛选 (Figure S8)。进而确定最佳的反应条件为:采用 [(MeCN)4Cu]BF4作为催化剂,TFA作为添加剂,在1,4-dioxane溶剂中反应,反应温度为125 oC,最终获得78%收率的内酯化产物C29。
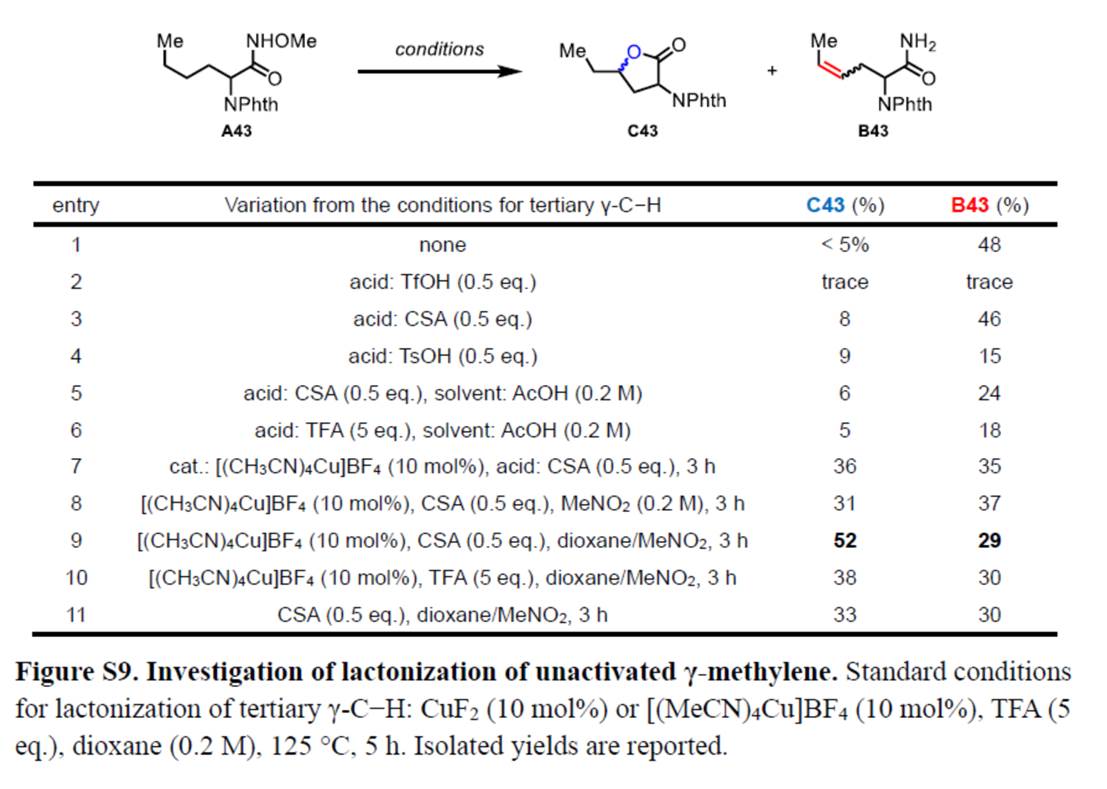
同时,作者还采用非活化N-甲氧基酰胺衍生物A43作为模型底物,进行相关内酯化反应条件的优化筛选 (Figure S9)。进而确定最佳的反应条件为:采用[(CH3CN)4Cu]BF4作为催化剂,CSA作为添加剂,在dioxane/MeNO2溶剂中反应,反应温度为125 oC,最终获得52%收率的内酯化产物C43。
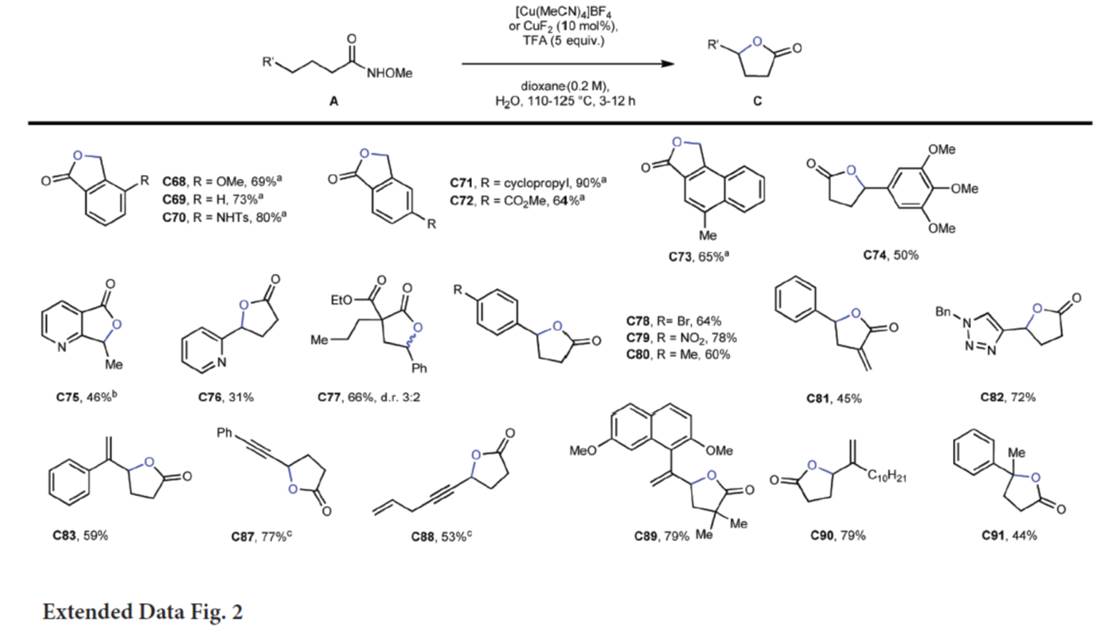
随后,作者对内酯化反应的底物 (Fig. 3与Extended Data Fig. 2)的应用范围进行深入研究。

之后,该小组通过如下的一系列研究进一步表明,这一全新的脱氢或内酯化策略具有潜在的实用性 (Fig. 4a与Fig. 4b)。同时,基于前期相关的文献报道[6],作者提出如下合理的反应机理 (Fig. 4c)。
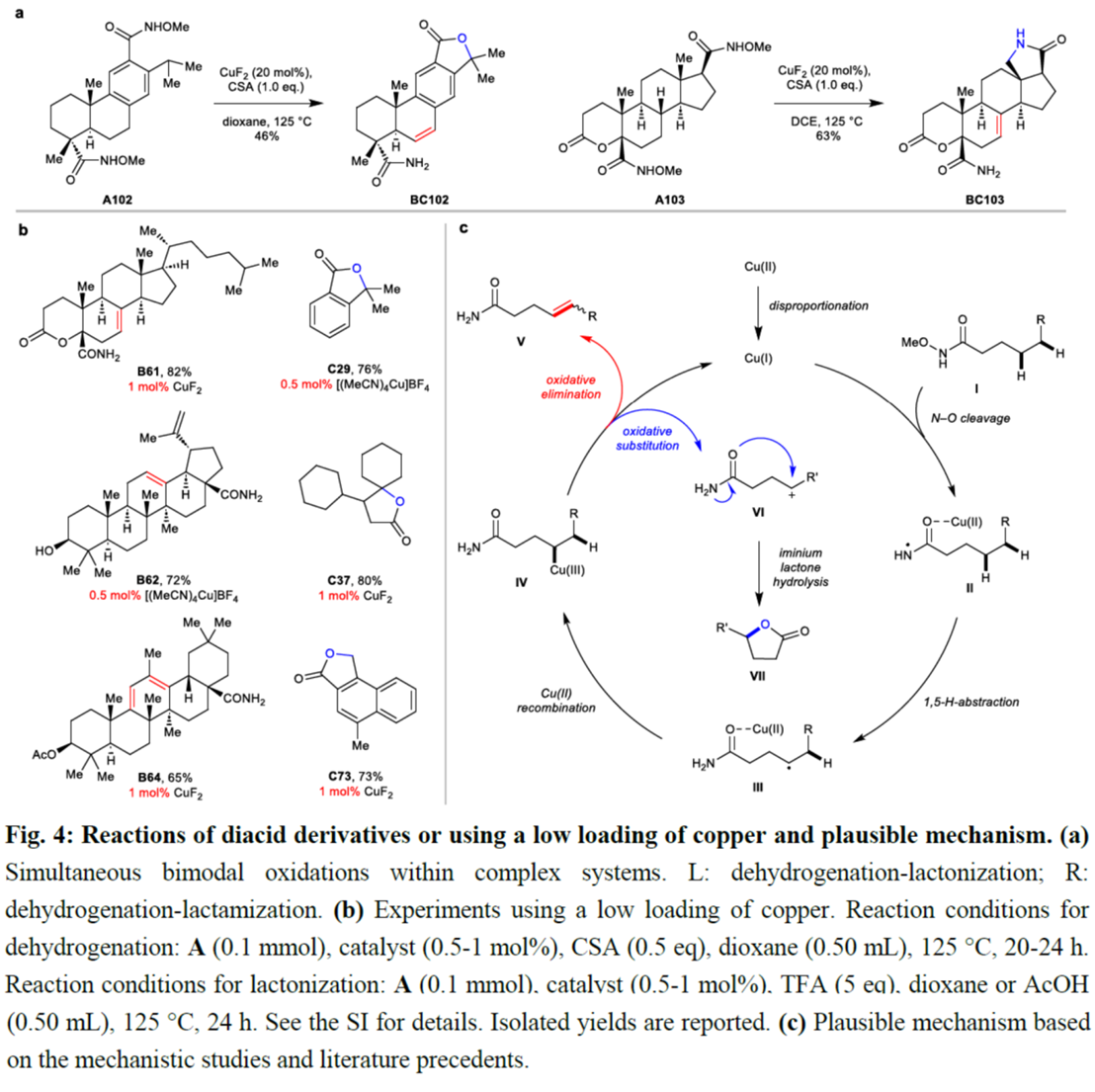
总结:美国Scripps研究所的余金权课题组报道一种全新的Cu(I)-催化N-甲氧基酰胺中γ-C(sp3)−H键的脱氢与内酯化反应方法学,进而成功完成一系列γ,δ-不饱和一级酰胺与γ-内酯分子的构建。这一全新的合成转化策略具有底物范围广泛、出色的官能团兼容性以及温和的反应条件等优势。
参考文献:
- [1] P. H. Buist, Nat. Prod. Rep. 2004, 21, 249. doi:10.1002/chin.200426285.
- [2] A. E. Rettie, A. W. Rettenmeier, W. N. Howald, T. A. Baillie, Science 1987, 235, 890. doi:10.1126/science.3101178.
- [3] R. Breslow, S. W. Baldwin, J. Am. Chem. Soc. 1970, 92, 732. doi:10.1021/ja00706a068.
- [4] D. Wang, M. Wasa, R. Giri, J. Yu, J. Am. Chem. Soc. 2008, 130, 7190. doi:10.1021/ja801355s.
- [5] A. Faulkner, N. J. Race, J. S. Scottb, J. F. Bower, Chem. Sci. 2014, 5, 2416. doi:10.1039/C4SC00652F.
- [6] A. L. J. Beckwith, A. A. Zavitsas, J. Am. Chem. Soc. 1986, 108, 8230. doi:10.1021/ja00286a020.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.













No comments yet.