
作者:石油醚
导读:
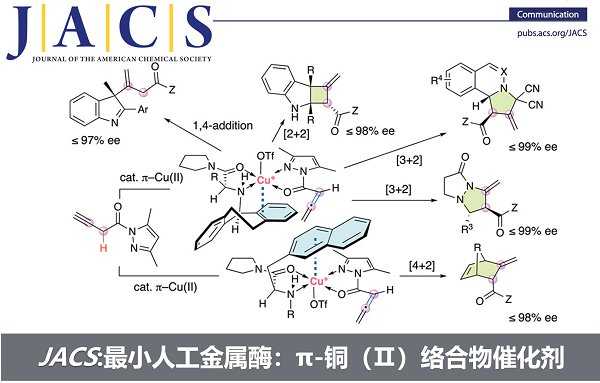
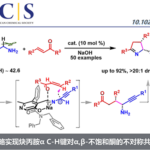
自2006年石原等人发表了关于π-铜(Ⅱ)络合物触媒的第一篇学术论文以来,这个领域的研究已取得了重要进展。氨基酸是酶蛋白的最小单元,我们课题组利用氨基酸衍生的单肽和铜(Ⅱ)盐的1:1配位络合物1或2作为催化剂,曾成功地促成了丙烯酰胺3和丙炔酰胺4的不对称环化加合反应,以及羧酸酰胺5的不对称α-卤代化反应和不对称α-胺化反应等(图1)。从化学合成的角度看,催化剂的分子量越小越有利,虽然其大小只有天然酶的百分之一,但作为不对称催化剂展现出了足够的功能,这点非常有趣。
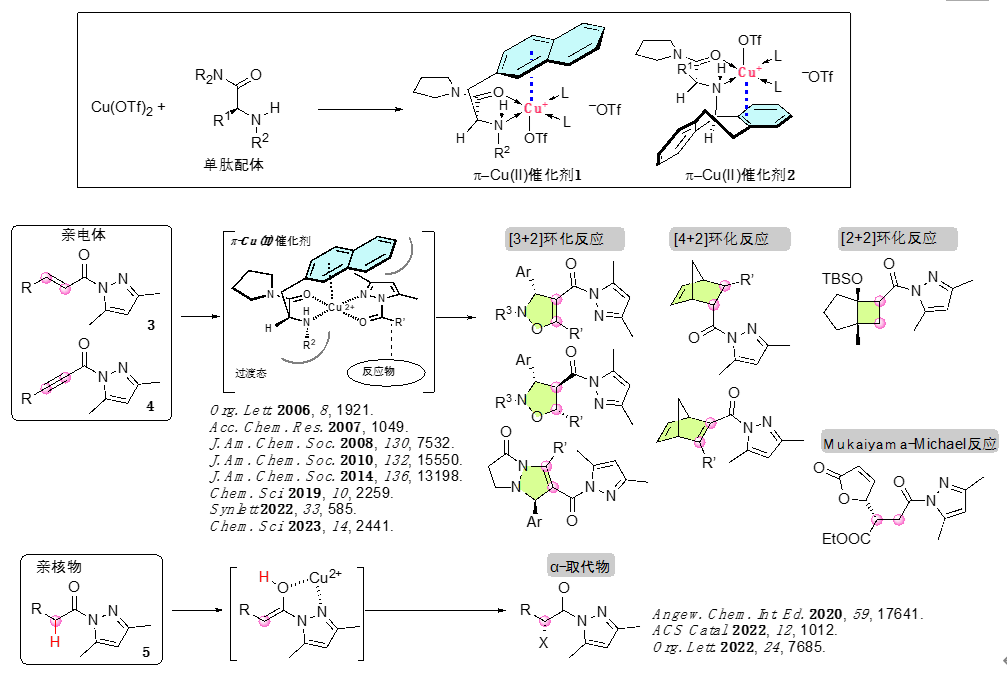 图1 Ishihara 等人开发的使用 π-铜(II) 催化剂的不对称反应示例
图1 Ishihara 等人开发的使用 π-铜(II) 催化剂的不对称反应示例
最近,石原团队在不对称催化领域迈出了新的步伐。我们创新地利用π-铜(Ⅱ)络合物作为不对称催化剂,原位生成难以分离的联烯酰胺,并成功地控制了其后续的α,β-位点选择性和对映选择性的Michael加成、[2+2]环加成、[3+2]环化加成和 [4+2]环加成(图2)。产物为光学活性的β,γ-不饱和羧酸衍生物,具有高附加值的复杂骨架,可用于探索光学活性药物和香料等领域。
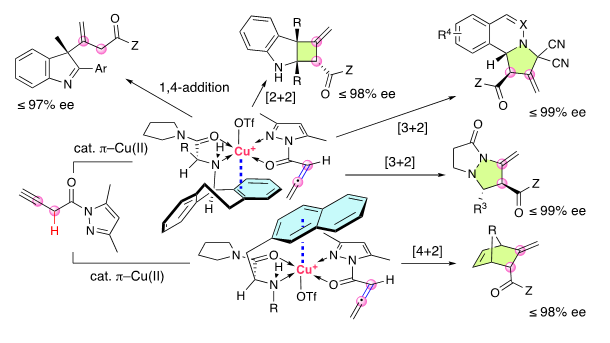 图2. 本工作
图2. 本工作
“Tandem Isomerization/α,β-Site-Selective and Enantioselective Addition Reactions of N-(3-Butynoyl)-3,5-dimethylpyrazole Induced by Chiral π–Cu(II) Catalysts.
Weiwei Guo, Masahiro Hori, Yoshihiro Ogura, Kazuki Nishimura, Kosuke Oki, Tomoyuki Ikai, Eiji Yashima, and Kazuaki Ishihara* J. Am. Chem. Soc. 2023, 145, 27080–27088. Doi: 10.1021/jacs.3c10820
前言
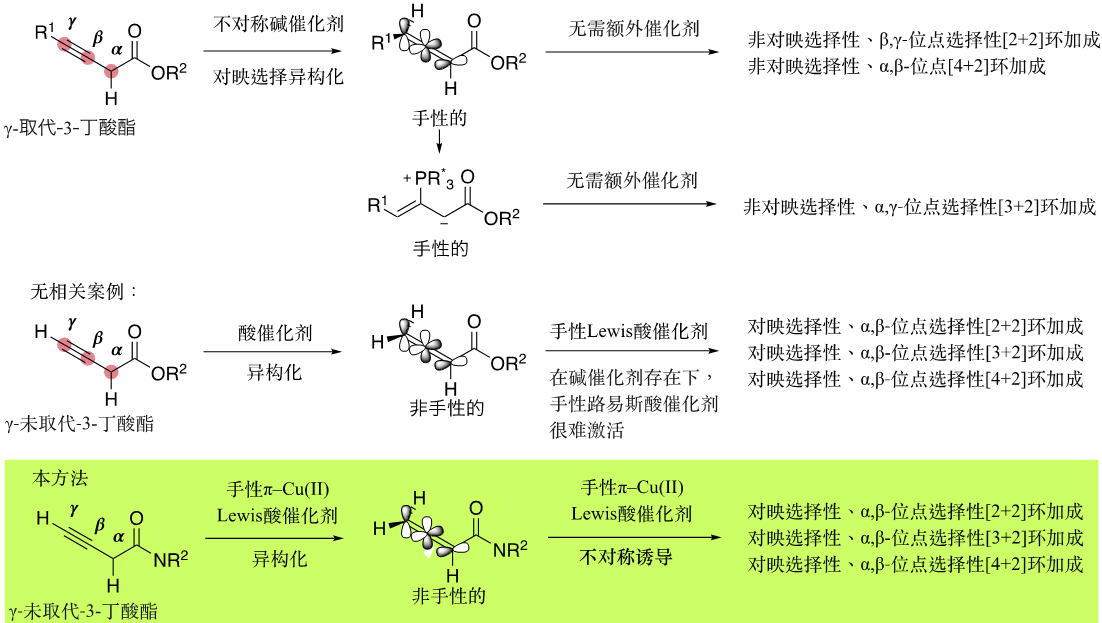 图3. 3-丁炔酸衍生物的串联不对称反应的历史背景
图3. 3-丁炔酸衍生物的串联不对称反应的历史背景
联烯在有机化学中非常灵活,因其具有两个垂直的π轨道,为多样化的合成提供了可能。其中,活化联烯的环加成反应是合成碳环和杂环化合物的有效方法。不对称版本的开发极大地扩展了其应用范围,特别是用于合成生物活性物质。近年来,人们开始研究3-炔酸酯作为潜在γ-取代活化联烯酯进行不对称环加成反应。γ-取代的联烯衍生物本身是手性的,可以通过使用手性碱催化剂对γ-取代-3-丁炔酸酯进行不对称异构化来合成(图3)。如果可以通过不对称合成得到γ-取代的联烯酯,其接下来的不对称[2+2]环化加合反应一般发生在其β,γ-位点。另一方面,对于其串联不对称[3+2]环化反应,一般Lewis碱催化剂会对其β位置亲核进攻,形成手性1,3-偶极体,进而得到α,γ-位置的环化产物。然而,γ-未取代的联烯衍生物本身是非手性的,几乎没有成功控制随后加合反应的对映选择性的例子。主要是因为不带γ-取代基团的潜在联烯的反应性、对映选择性、立体选择性和位点选择性很难控制,这是由于它们的不稳定性、多个反应位点和结构简单性造成的。此外,如果Lewis得以催化异构化合成联烯酯类衍生物,那么同时该联烯就很有可能被Lewis酸活化,进而实现其串联手性和α, β-位点选择性的环加成反应。
根据我们之前的研究,Cu(II)与N-酰基吡唑之间的螯合作用能显著增加羰基部分 α-质子的酸性。 根据B3LYP/6-31+G*理论水平的DFT计算结果,N-(3-丁炔酰基)-3,5-二甲基吡唑1的伪-Z构象的MEP值为157.3 kJ/mol,相应的pKa为17.0。 这个pKa值远低于丙酮(pKa = 26.5)和叔丁基乙酸酯(pKa = 20.3)的值(图4)。 基于这些结果,我们推测1和π-Cu(II)催化剂配位后Ha相对更高的酸性可能导致1异构化,生成相应联烯酰胺2。此联烯酰胺会被π-Cu(II)催化剂进一步活化,从而能被亲核试剂直接捕捉而发生反应。此时的π-Cu(II)催化剂起到三个作用:第一,催化潜在联烯1的原位异构化;第二,活化联烯发生后续串联反应;
第三:创造手性环境。(图5)
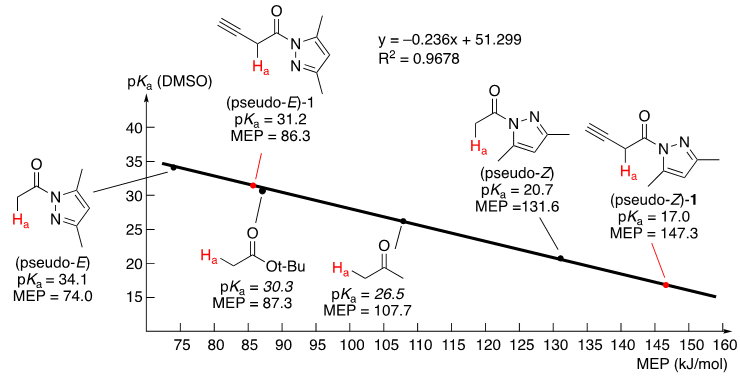
图4. 1和其相关化合物的MEP 及pKa 值:The linear relationship between pKa (DMSO) and MEP values. Calculated pKa values: plain numbers. Measured pKa values: Italic numbers. The equation (y = –0.236x + 51.299) was calculated based on the pKa of known compounds and MEP values of these compounds calculated by us.
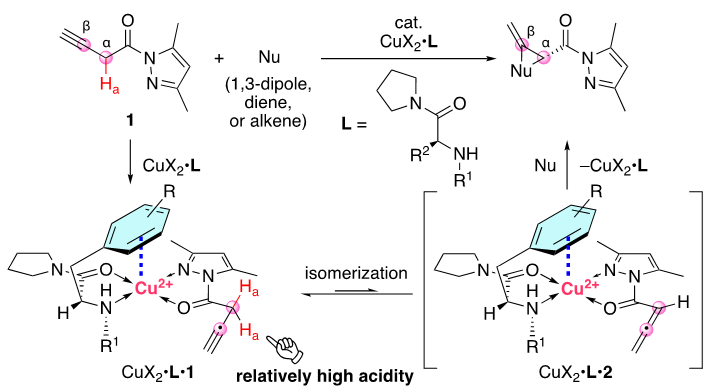
图5. CuX2•L-catalyzed α,β-Site- and Enantioselective Cycloaddition Reactions of In-situ-generated N-Allenoyl-3,5-dimethylpyrazole 2 with Nu
Part1:潜在联烯1与偶氮亚胺3的α,β-位点/对映选择性[3+2]环加成反应
为了验证我们的猜想,我们以二氯甲烷为溶剂在-40°C 下,对N-(3-丁炔酰基)-3,5-二甲基吡唑1和偶氮甲烷亚胺3a进行了α,β位选择性和对映选择性的 1,3-偶极环加成的研究。我们首先对配体进行了筛选,确定了L6是最佳配体。有意思的是筛选过程中发现即使是同一构型的手性配体(s)-L,使用不同的N-取代基却会生成绝对构型相反的产物,比如用小位阻的甲基(Table 1),生成的产物主要构型是5S,6R(entry 1),而用更大位阻或者Dbs时产物的绝对构型发生了反转,生成的是5R, 6S构型(entry 4-8)。这意味着,使用不同的N-取代基配体,反应过程中手性环境并不相同。为了理解这些结果,首先我们对异构化产物联烯的构型进行了理论计算,发现其畸变构象的cis-2(约40°)最稳定(图7,左)。其次当我们使用非芳香的环己烷取代的L8配体,我们发现产物仍旧有高达93%的ee值(entry 8)。鉴于这些并结合之前的研究,我们提出了以下反应历程:首先,Cu(OTf)2L•1更倾向于L的氮原子和1的氮原子之间的反式螯合,这是基于对Cu(NTf2)2•(S)-L2•2-乙酰基吡啶的CD分析观察到的正Cotton效应(见SI)。对于Cu(OTf)2•L1(图7,右,TS-A1和endo-TS-A2),L1配体的2-萘基部分与Cu(II)之间的π−Cu(II)相互作用会封锁2的的si-面,这将导致偶氮亚胺3a进攻re-面,从而生成(5S,6R)-4a作为主要产物。但N-R1的位阻增大会阻止3a接近re-面(entry1-entry 5)。在entry4-entry5中,由于N-R1阻止了re-面,而不是π−Cu(II)相互作用阻止了si-面,因此观察到了相反的对映选择性。对于Cu(OTf)2•L6而言,此时不是2-萘基,而是N-Dbs结构中的苯环与Cu(II)产生了π−Cu(II)相互作用,封锁2的si-面,形成了与之前相反的手性环境(图7,右, TS-B1和endo-TS-B2)
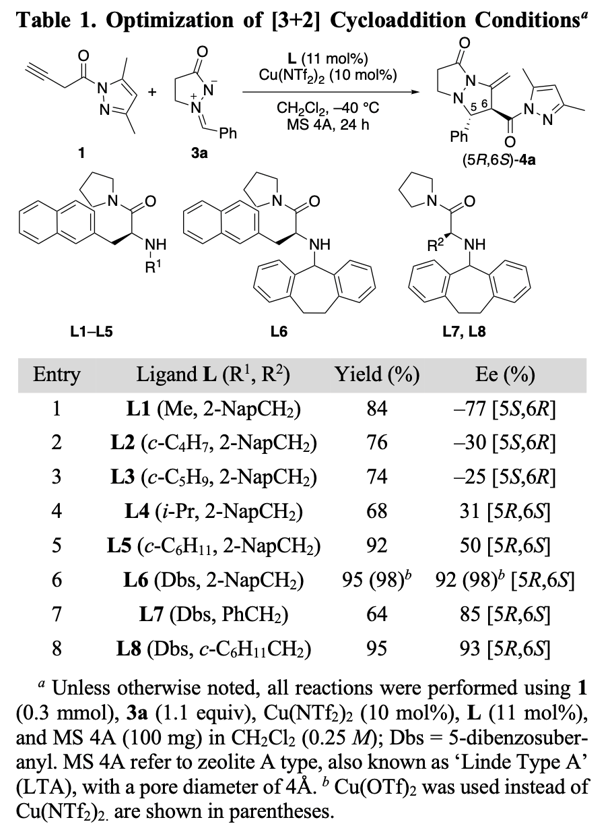
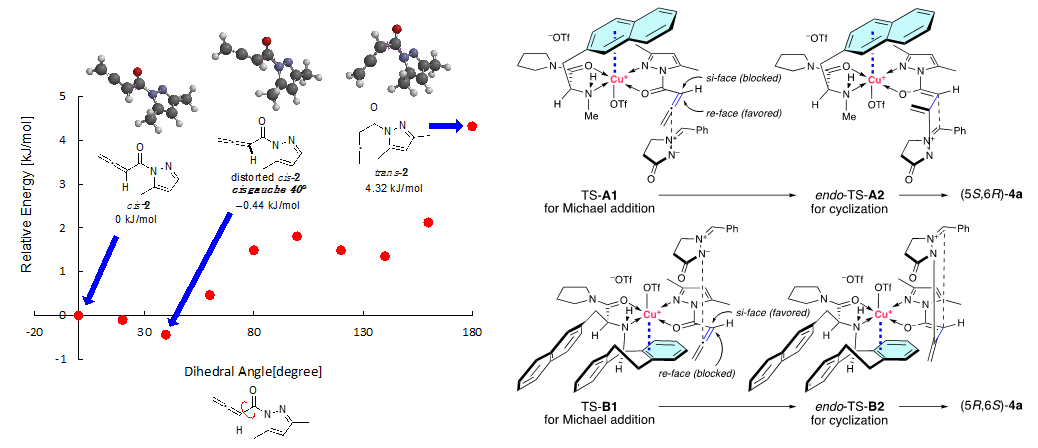
图7. 左:2的二面角与其相对能量之间的关系;右:提出的Cu(OTf)2·L1 和 Cu(OTf)2·L6 催化的 1 与 3a 的 [3 + 2] 环加成反应的过渡态模型
利用L6作为π–Cu(II)路易斯酸催化剂的手性配体,我们进一步评估了该串联不对成[3+2]环加成反应的普适性。苯环上带有吸电子基和供电子基的偶氮亚胺3都能兼容,且都能以高收率高对映选择性的得到产物(图8)。此外,苯环的对位或间位取代基(4a–k, 4l–o, 4s, 4t)也均能兼容。然而,含有邻溴取代基的产物4q的对映选择性略有降低(78% ee)。这可能归因于邻位溴取代基的立体位阻。含有2-萘基的偶氮亚胺3r也能被兼容,以90%收率,98%的ee值得到产物4r。我们发现,反应对含杂环和苯乙烯基的底物也是适用的,均以高产率和高对映选择性获得所需产物(4u: 98%,94% ee;4v: 96%,92% ee)
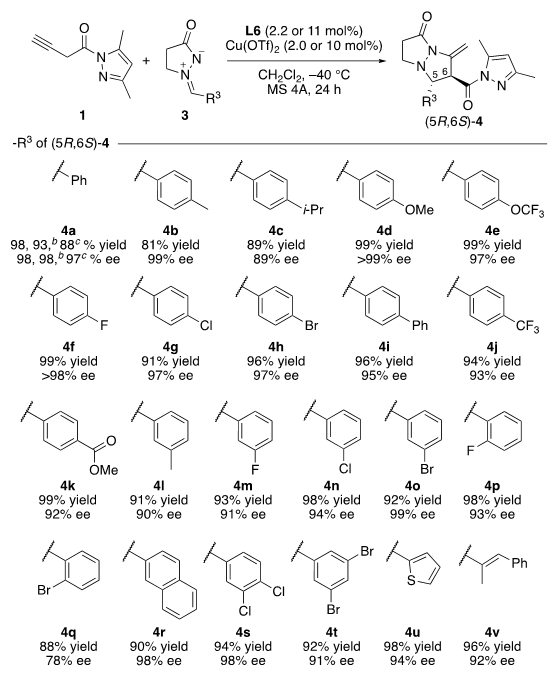
图8. 潜在联烯1与偶氮亚胺3的α,β-位点/对映选择性[3+2]环加成反应
为展示该串联反应的实用性,我们随即进行了克级反应,且催化剂当量降低至5%mol,产物能以96%的收率,95%的ee值得到。(图9)此外,配体L6可以以96%的收率回收,适用于环保的方案。而且,4a的转化提供了增加分子多样性的机会。例如,双键轻松异构化得到了5a,收率为98%。还原和醇解可分别得到6a和7a,收率分别为85%和90%。
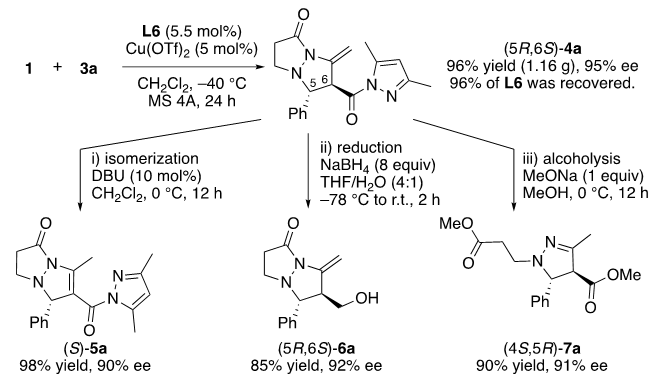
图9. 克级反应和产物转化
Part2:异喹啉叶立德8与1进行串联异构化/α,β-位点和不对称去芳构化 [3+2] 环加成反应
近年来,异喹啉叶立德的不对称1,3-偶极环加成反应吸引了科学界的广泛关注。这源于它们在构建吡咯异喹啉骨架方面的方面的高效性,这些骨架是许多天然产物和药用活性化合物中至关重要的组成部分。2015年,Waldmann团队首次报道了使用异喹啉叶立德与联烯酸酯进行区域选择性的去芳构化 [3+2] 环化反应。随后,Guo团队进一步扩展了其底物范围至结构相似的酞嗪叶立德。值得注意的是,这两种催化方法生成的都是β,γ-选择性的产物。但至今尚未开发出有效的不对称版本,尽管已经做了专门的尝试。因此,我们随后尝试了异喹啉叶立德8与1进行串联异构化/α,β-位点不对称去芳构化 [3+2] 环加成反应。反应的最初优化见SI,section 5,我们最终选择了L9作为手性配体。如图10所示,含有不同取代基的异喹啉叶立德8可以有效地与1发生反应,在大多数情况下都以高产率和高对映选择性地得到exo-产物9(对映选择性高达>99% ee)。值得注意的是,带有给电子基的8f也表现良好(9f:97% 收率,91% ee)。使用酞嗪叶立德(X = “N”)作为偶极子,反应也能顺利进行,以93%的产率得到对映体纯度高达>99% ee的9g。
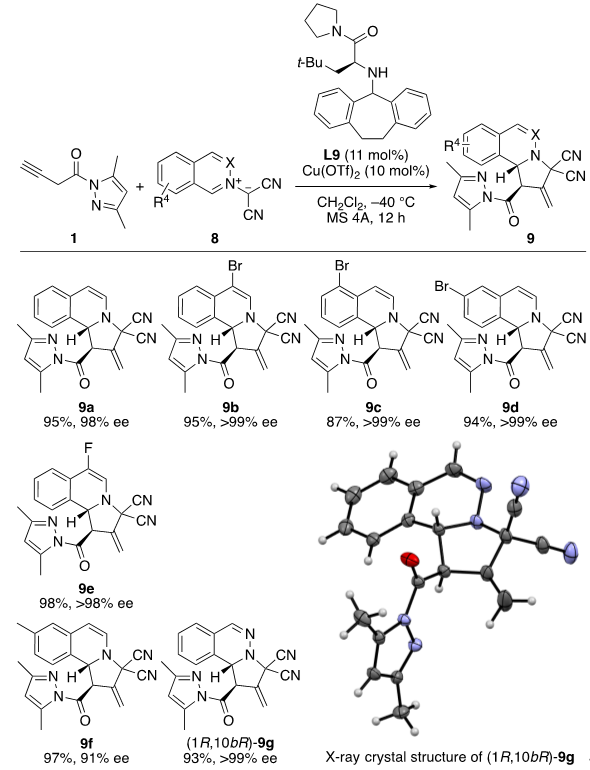
图10. 异喹啉叶立德8与1进行串联异构化/α,β-位点和不对称去芳构化 [3+2] 环加成反应
Part3:潜在联烯1与共轭二烯10的α,β-位/对映选择性 Diels-Alder 反应
Diels–Alder反应是有机化学中最强大的反应之一,因为它允许在单步反应中形成两个C–C键并构建多达四个相邻的立体中心。在过去几十年中,对活化烯烃的催化不对称Diels–Alder反应的研究越来越深入。然而,以活化联烯作为亲双烯体的报道却非常少,尽管其在天然产物的全合成中具有潜在的实用价值。其中一个原因可能是很难获得合适的活化联烯(例如,在laurenditerpenol的全合成中,Corey团队使用三氟乙基联烯酯作为亲双烯体,但此联烯酯的合成需要四步且总产率仅为2%)。因此,我们接下来调研了原位生成联烯酰胺2的策略用于串联[4+2]反应的可行性。
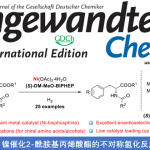
如图11所示,以DCM为溶剂,我们首先使用L6(11 mol%)和Cu(OTf)2(10 mol%)对1与环戊二烯10a进行串联异构化/α,β-位点和手性选择性[4+2]环加成反应进行了研究(entry1)。尽管11a以42%的产率得到,但其endo选择性和对映选择性仅为中等。幸运的是,当使用L2代替L6时,11a的产率提高至83%,且其endo选择性和对映选择性也显著提高至96%endo和98% ee(entry 2)
随即我们展开了反应普适性的调查。如表3所示,1与环状二烯(例如取代环戊二烯,富烯和呋喃)的[4+2]环加成反应能高效进行,以高收率,高endo选择性和高对映选择性的得到产物11。值得注意的是,即使用立体位阻更大的10b和10d,该串联反应仍顺利进行,以高收率高、endo选择性和高对映选择的得到11b和11d(entry 3和5)。令我们感到惊喜的是,呋喃10e和10f也可高效反应(entry6和7)。呋喃最近在[4+2]环加成反应中受到特别关注,因为它们是至关重要的可再生平台分子,具有更可持续地生产化学建筑基块和材料的巨大潜力。与此相反,2与非环二烯的反应没有产生所需的环加成产物,可能是因为2的不稳定性和非环二烯的低反应性。
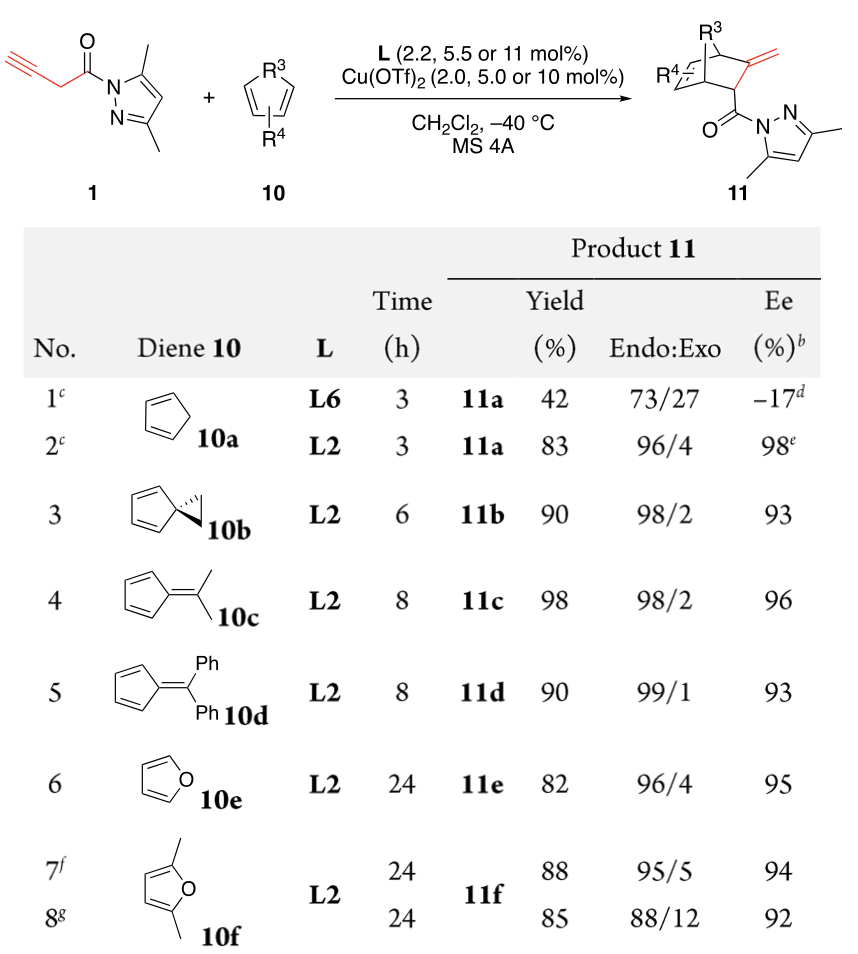
图11. 潜在联烯1与共轭二烯10的α,β-位/对映选择性 Diels-Alder 反应
我们随后对产物11进行了进一步转化,如图12所示。11a的乙醇解可以80%的收率生成相应的酯12a,其可用于合成天然产物(−)-β-santalene 的对映异构体。使用NaBH4还原11f可以以91%的收率得到13f,其是合成生物活性天然产物(−)-laurenditerpenol对映异构体的中间体。
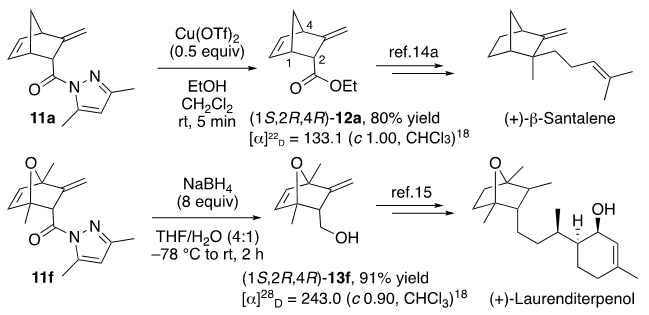
图12. [4+2]产物的转化
值得注意的是,由于其不定性,2无法被分离得到,但通过获得其加成产物,可以确认2的原位生成。此外,在标准反应下,仅仅使用配体无法得到异构化产物。但只使用Cu(OTf)2,不使用配体,[4+2]反应能顺利进行,这些控制实验说明了Cu(II)对1的原位异构化是至关重要的。
Part4:潜在联烯1与2,3-二取代吲哚的α,β-位点/不对成去芳构化1,4-加成和[2 + 2]环加成。
为了展示当前对1的催化异构化的更广泛应用,我们测试了2,3-二取代吲哚和原位生成的联烯酰胺2的亲核加成反应。如图13所示,2-芳基-3-甲基吲哚14a,14b与1在标准条件下可以高效的反应,得到含有全碳季碳中心的去芳构化产物15a(87%,97% ee)和15b(84%, 96% ee)。有趣的是,将吲哚的2号位换成烷基,后续可在联烯酰胺2的α位发生环化,生成含有三个手性中心,两个全碳季碳中心的去芳构化[2+2]环化产物16c-16f (ee:93%-98%)。
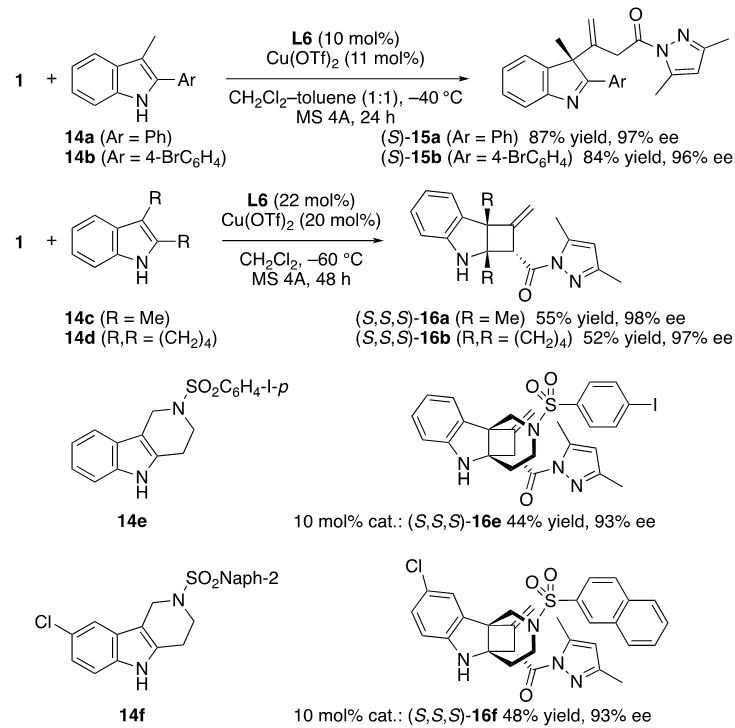
图13. 1和14的串联异构化/α,β位点选择性和对映选择性去芳构化 1,4-加成或 [2 + 2] 环加成反应
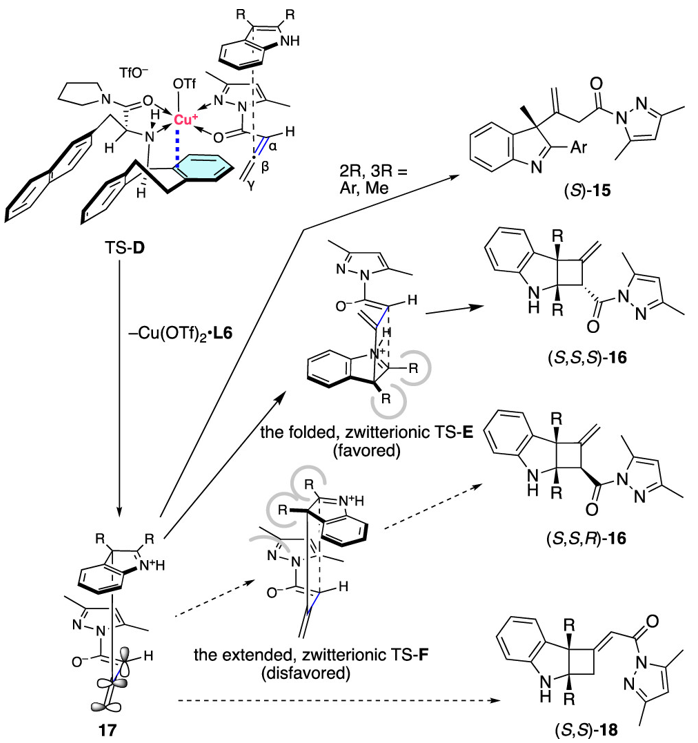
图14. 反应机理研究
我们可以合理地解释,2,3-二取代吲哚14对1的α,β-位点选择性不对称1,4-加成是通过类似于TS-B的TS-D进行的,生成了共同中间体17(图14)。当使用2-芳基-3-甲基吲哚时,由于2-芳基基团的立体位阻和/或亚胺离子与2-芳基基团的共轭稳定性,随后的环化反应受到抑制(参考SI,section 7)。因此, (S)-15成为主要产物。相反,使用2,3-二烷基吲哚时,后续的环化反应以α-位点选择性进行(>99% 形成于2的α-位点),并且对立体选择性也很高,通过折叠的、离子态的TS-E形成[2 + 2]环加成物(S,S,S)-16。然而,另一种立体异构体(S,S,R)-16则无法通过TS-F形成,因为2,3-二烷基基团与吡唑酰基团之间的立体位阻。有趣的是,通过17进行的γ-位点选择性环化反应以形成(S,S)-18并未发生。与此相反的是,目前仅有少数关于吲哚14与富电子联烯氨发生β,γ-位点选择性去芳构化(环)加成反应的例子。此外,已报道的热力学上烯烃与缺电子联烯酸酯的[2 + 2]环加成反应是通过协同的或异步的[π2s + (π2s + π2s)]机制发生在β,γ-位点。因此,图13中展示的α,β-位点选择性反应是非常罕见和有价值的结果。
总结
我们利用了被视为最小人工金属酶的π-铜(Ⅱ)络合物催化剂,成功地控制了结构不稳定的联烯衍生物的不对称反应。利用了联烯衍生物的合成前体,即3-丁炔酸酰胺,通过单步反应解决了这一挑战。关键在于使用了3-丁炔酸和3,5-二甲基吡嗪的酰胺作为原料。这样做可以增强相应酰胺的α位酸性,并且只通过π-铜(Ⅱ)络合物催化剂的作用,就能在一个反应釜中控制异构化和随后的不对称加成反应。此外,通过对天然来源的氨基酸进行化学修饰,也可以区分出两种对映异构体。最后,我们想强调这些成果是基于自2002年开始π-铜(Ⅱ)络合物催化剂研究以来,持续22年积累的众多发现上取得的成就。
(Kazuaki Ishihara教授供稿)
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.












No comments yet.