

本文作者:杉杉
导读
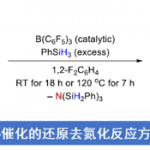
近日,Columbia大学的T. Rovis课题组在J. Am. Chem. Soc.中发表论文,报道一种通过镍/光氧化还原双重催化策略,进而成功实现一系列三烷基胺化合物的N-甲基选择性α-芳基化反应。这一全新的双重催化策略具有温和的反应条件、优良的 N-甲基选择性以及良好的底物应用范围等优势。并且,上述策略同样能够进一步应用于一系列药物分子的后期芳基化过程。同时,作者通过对反应机理的实验研究表明,Ni催化剂在捕获α-氨基自由基时,表现出较为独特的反应活性,仅有一级α-氨基自由基能够成功完成相应的交叉偶联过程。
Late-Stage N‑Me Selective Arylation of Trialkylamines Enabled by Ni/Photoredox Dual Catalysis
Y. Shen, T. Rovis, J. Am. Chem. Soc. 2021, ASAP. doi:10.1021/jacs.1c08157.
正文
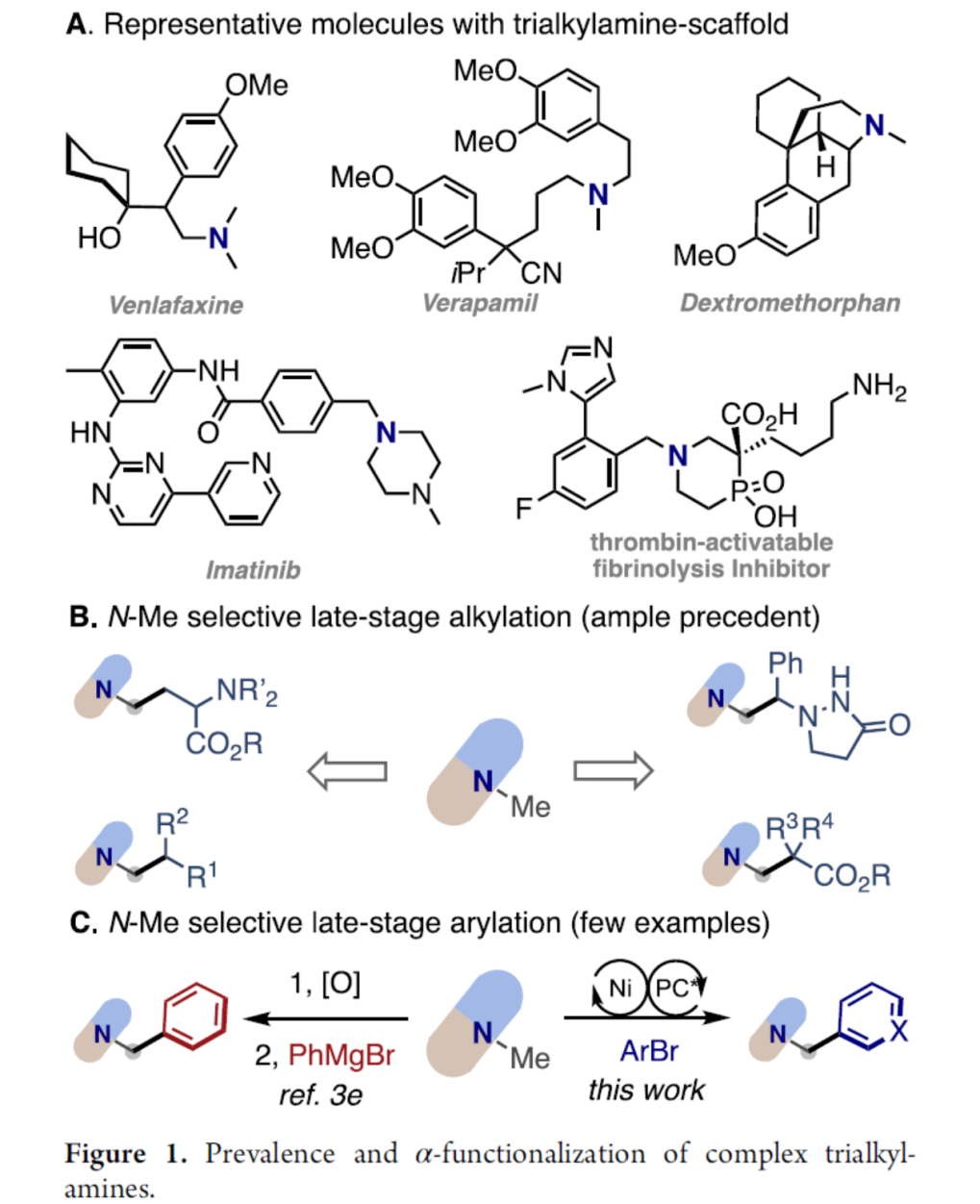
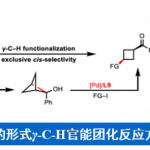
三烷基胺结构单元广泛存在于一系列生物碱类天然产物、农用化学品、临床药物分子以及各类市售药物中 (Figure 1A)。其中,通过C-H键α–位置到N-位置的官能团化反应方法学,能够有效地实现相关有机分子物理特性、生物活性以及药代动力学活性等方面的精细调节[1]。同时,复杂三烷基胺化合物直接的后期α-C(sp3)-H键官能团化反应方法学相关研究的进一步发展,已经成功设计出一系列全新的合成切断模式,进而能够加速各类新型先导化合物的发现。并且,迄今为止,对于复杂三烷基胺底物后期α-烷基化方法学的研究,同样已经取得诸多的研究进展[2] (Figure 1B)。然而,对于三烷基胺底物芳基化反应策略的设计,则仍有待进一步研究[2e] (Figure 1C, left)。
近期,过渡金属/光氧化还原双重催化策略,已经为在较为温和的反应条件下,实现一系列在有机合成化学中具有巨大挑战性的相关化学键的构建,开辟出一条全新的反应途径[3]。同时,由于三烷基胺在一系列过渡金属/光氧化还原催化方法学的研究中,通常作为一种通用的氢负离子源、氢源或电子源,并由此使三烷基胺的直接芳基化过程无法与上述的相关转化过程进行有效的竞争[4]。然而,Molander课题组在对于α-硅基三烷基胺的去硅基芳基化方法学 (desilylative arylation)的开创性研究中发现,通过在二级胺分子中预先引入硅基基团,能够有效地实现α-氨基自由基形成过程中的区域选择性控制[5]–[6]。由此,作者设想,能否设计一种较为通用的反应策略,实现一系列三烷基胺分子的区域选择性芳基化过程,进而成功完成各类新型苄基二级烷基胺分子的制备 (Figure 1C, right)。基于上述设想,本文作者报道一种通过镍/光氧化还原双重催化的反应策略,进而成功实现一系列三烷基胺化合物的N-甲基选择性α-芳基化过程。
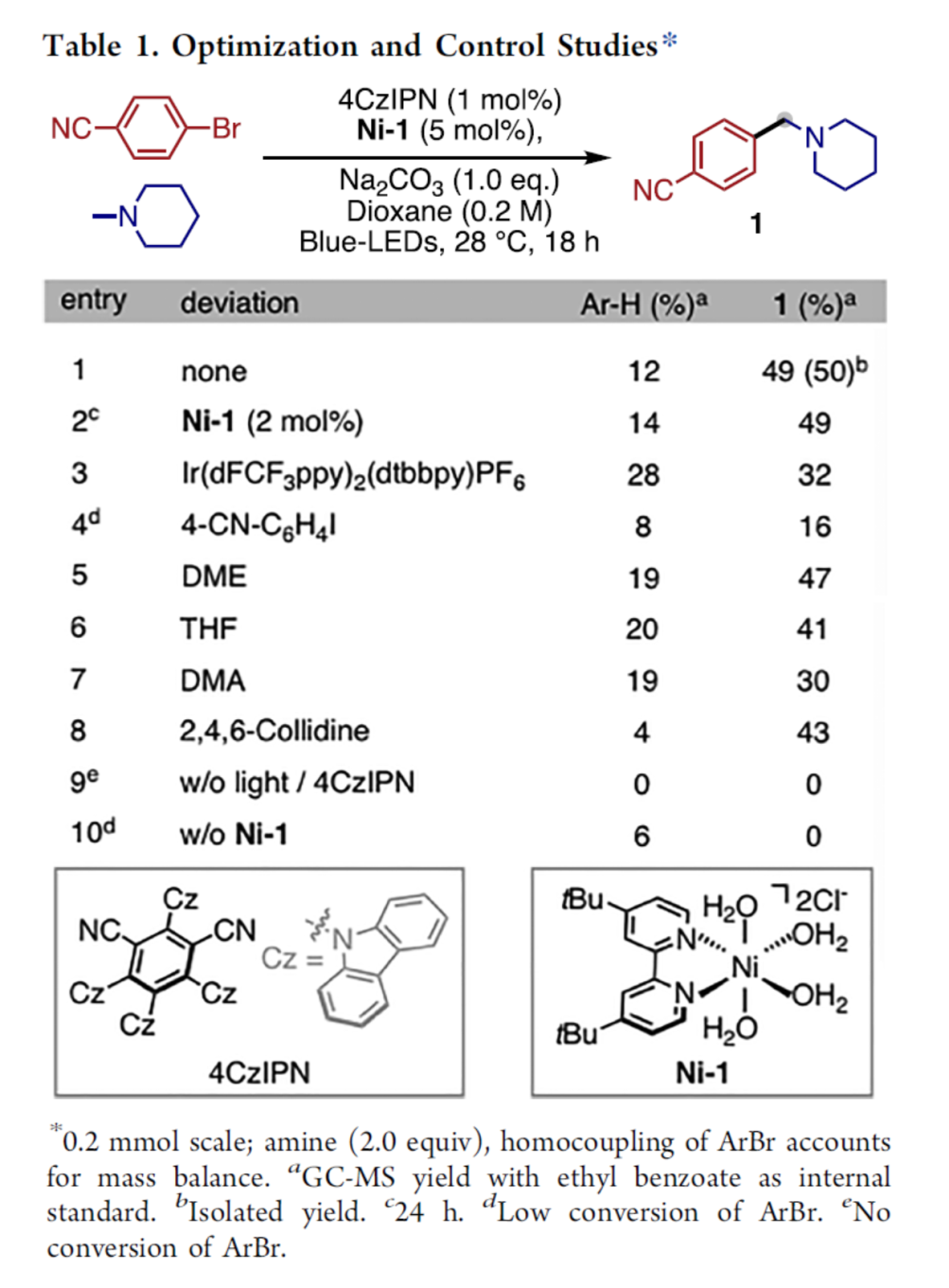
首先,作者采用4-溴苯腈与N-甲基哌啶作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用4CzIPN作为光催化剂,NiCl2(dtbbpy)(H2O)4 (Ni-1)作为镍催化剂,Na2CO3作为碱,在二氧六环反应溶剂以及蓝光LED辐射 (440 nm)的条件下进行反应,最终获得50%收率的N-甲基芳基化产物1。
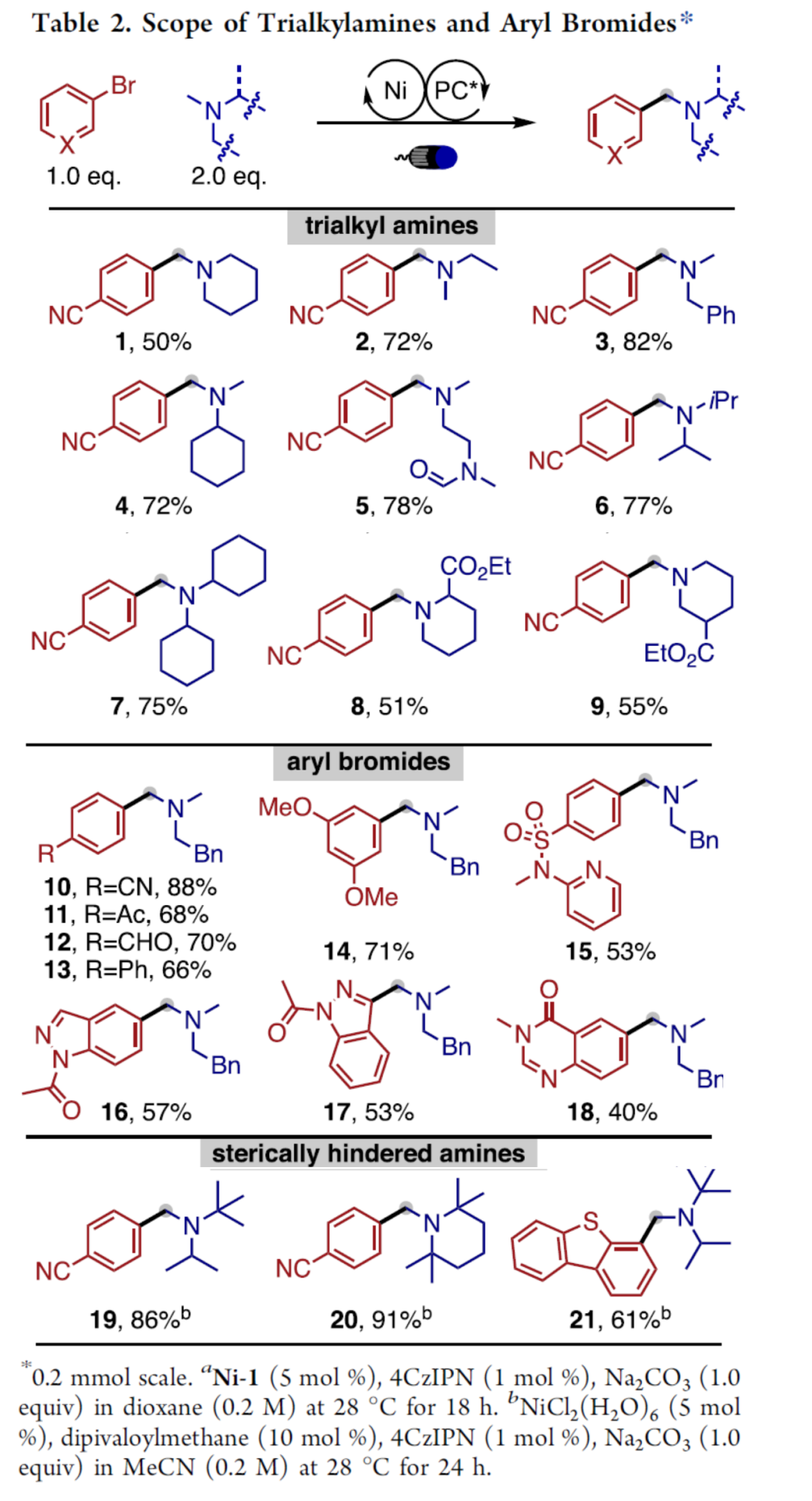
在上述的最佳反应条件下,作者对相关底物的应用范围进行考察 (Table 2)。研究表明,这一全新的α-芳基化策略具有优良的N-甲基选择性。各类具有不同烷基取代的三烷基胺底物,均能够顺利地与4-溴苯腈进行反应,并获得预期的芳基化产物1–9,收率为50-82%。之后,该小组发现,一系列具有不同官能团以及杂环基团取代的芳基溴底物,同样能够顺利参与上述的转化过程,进而获得相应的芳基化产物10–18以及21,收率为40-88%。值得注意的是,上述的标准反应条件,对于底物中存在的酮羰基与醛羰基官能团,均能够有效地兼容。此外,作者进一步观察到,在镍催化剂以及1,3-二酮配体存在的条件下,具有更高立体位阻的三烷基胺底物,同样能够良好地兼容,并获得相应的目标产物19–21,收率为61-91%。
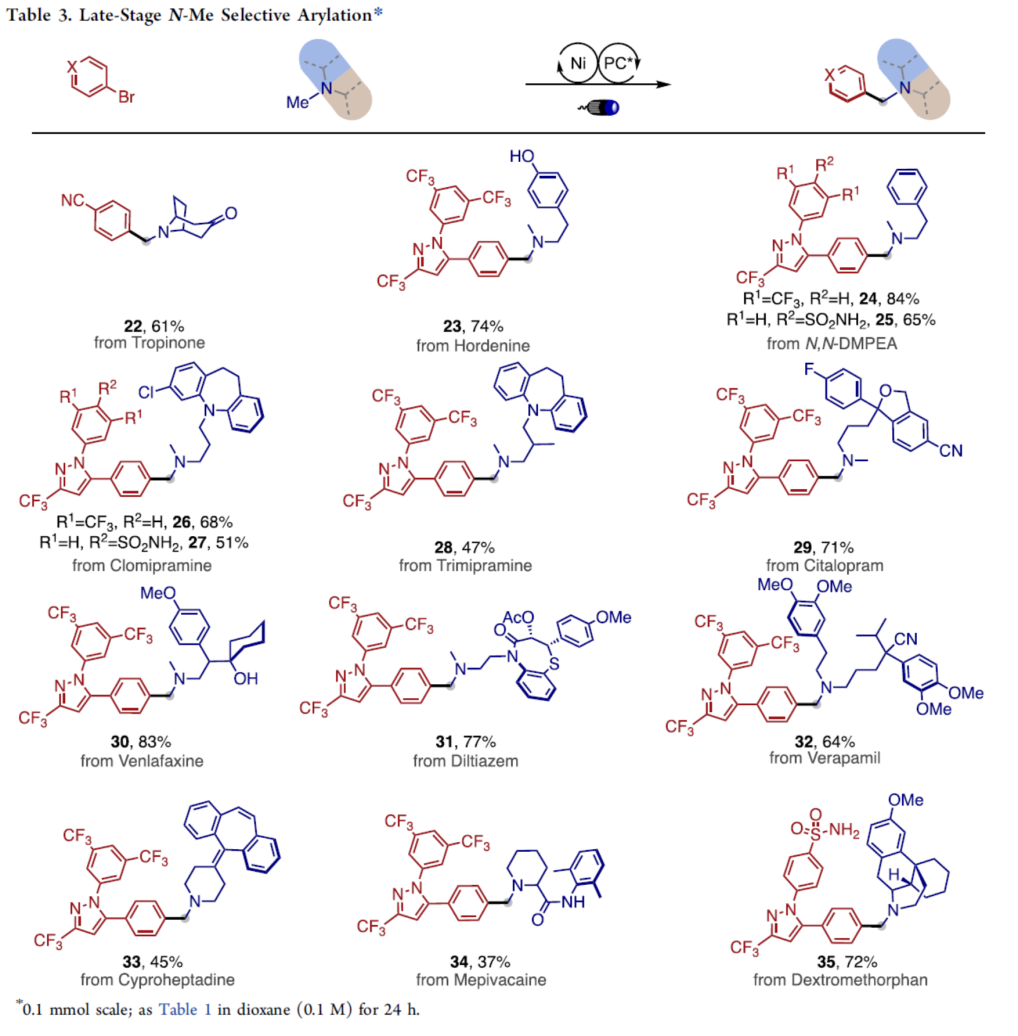
为进一步研究上述α-芳基化方法学的合成应用价值,接下来,作者对一系列具有三烷基胺结构单元的药物与天然产物分子进行相关的后期官能团化反应研究 (Table 3)。实验表明,通过这一全新的N-甲基选择性α-芳基化策略,能够顺利完成一系列通过常规策略难以获得,并具有一定药学应用价值的苄基二级烷基胺分子22–35的构建 (37-84% 收率)。
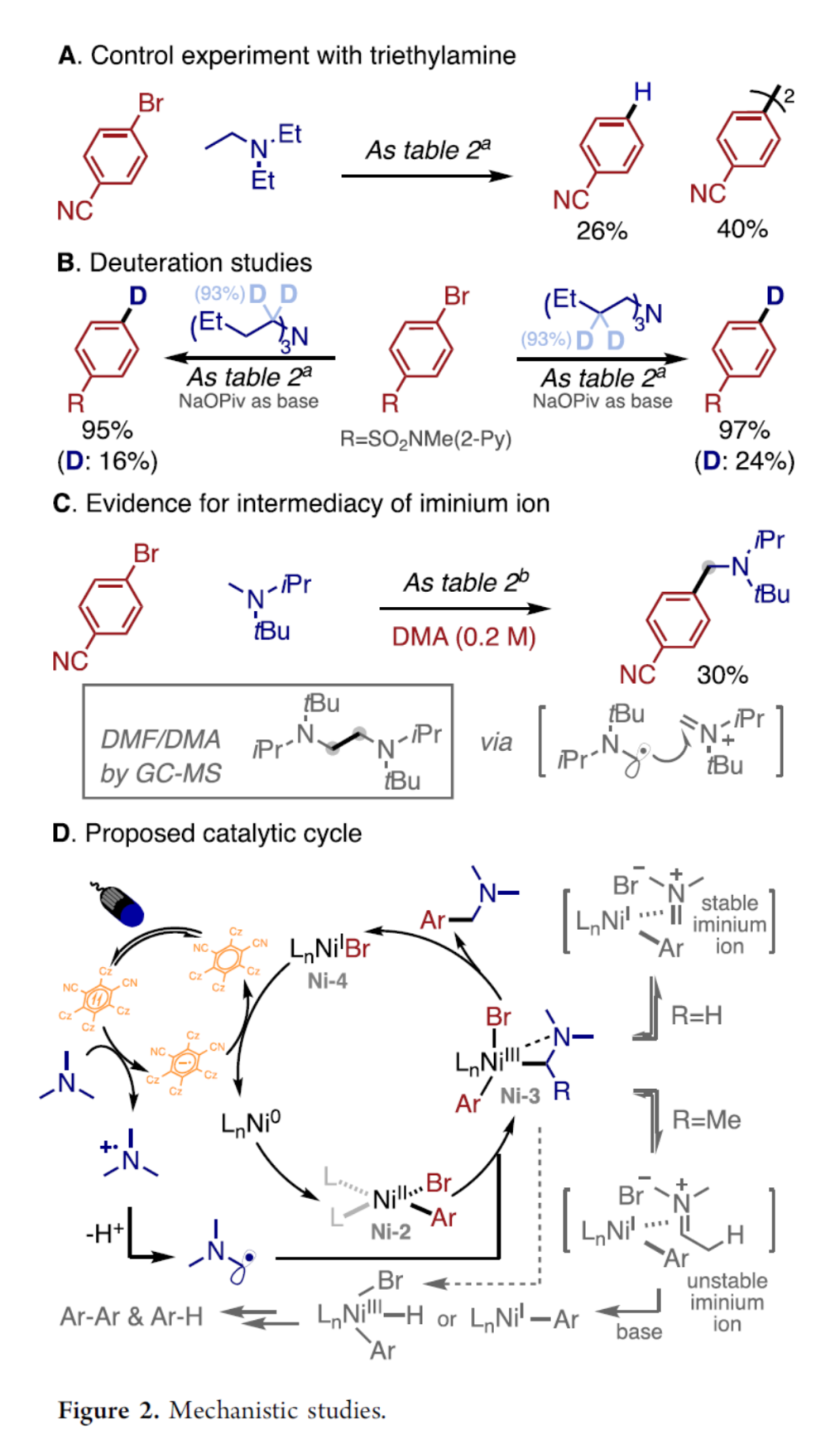
通常,通过三烷基胺的氧化/去质子化过程,优先形成具有较低立体位阻的α-氨基自由基[6a], [6b], [7a]。然而,这一反应路径无法完全解释在上述光化学芳基化过程中观察到的较为独特的N-甲基选择性,尤其对于能够通过氧化/去质子化串联过程,形成具有中等选择性比率的α-氨基自由基混合物 (例如N-甲基哌啶底物,在相应氧化/去质子化的反应条件下,能够形成具有2:1选择性比率的一级与二级α-氨基自由基混合物)的相关底物[6a], [6c]。之后,该小组对于反应过程中,区域选择性的相关控制因素进行深入研究。首先,作者通过在上述标准反应条件下,选择三乙胺底物进行的控制实验研究中发现,仅能够检测出26%收率的加氢去溴化(hydrodebromination)产物以及40%收率的同二聚 (homodimerization)产物,并未获得预期的芳基化产物。通过这一实验观察,能够排除ArNiIILnBr (Ni-2)中间体捕获二级α-氨基自由基的机理路径 (Figure 2A)。之后,作者选择α-与β-氘代三丁胺底物进行的氘代实验研究中观察到,加氢去溴过程的氢源,可能源自于三丁胺分子α-与β-C(sp3)-H键以及溶剂 (二氧六环)分子 (Figure 2B)。同时,在对于具有显著立体位阻的三烷基胺底物芳基化反应条件的优化过程中,作者发现,在极性非质子溶剂 (DMF或DMA)中,能够观察到胺底物较为显著的二聚过程,这可能源自于氨基自由基对于亚胺离子中间体的进攻 (Figure 2C)。同时,研究发现,在N-甲基哌啶的标准芳基化反应体系中,加入H2O (50 eq.)时,反应过程受到抑制,仅获得相应的加氢去溴化产物,相关过程可能涉及亚胺离子中间体的水解。基于上述实验观察以及文献中对于镍配合物与亚胺离子之间配位过程的相关研究报道[8]–[9],作者提出一种可能的反应机理 (Figure 2D)。首先,通过氧化加成配合物 Ni-2对于α-氨基自由基的捕获过程,形成NiIII中间体 (Ni-3),并且,反应过程中同样可能经历亚胺离子与Ni-4中间体之间的非循环平衡过程 (off-cycle equilibrium)。之后,通过Ni-3中间体的还原消除过程,形成相应的芳基化产物。同时,研究发现,具有β-H的亚胺离子则容易使Ni-3中间体出现分解,并产生相应芳基溴底物的加氢去溴化以及同二聚的副产物。并且,上述的实验观察与Pierpont以及Barefield团队报道的相关(R3P)2Ni(0)/亚胺离子配合物的稳定性相一致[9]。
总结
Columbia大学T. Rovis课题组报道一种采用镍/光氧化还原双重催化的设计策略,进而能够在较为温和的反应条件下,成功实现一系列具有三烷基胺结构单元的药物分子的后期芳基化。同时,上述反应策略对于N-甲基中的C(sp3)-H键表现出极为优良的选择性,不仅能够极大地简化苄基二烷基胺分子的合成路线,并且,同样能够加速相关先导分子的发现。
参考文献
[1] M. A. A. Endoma-Arias, D. P. Cox, T. Hudlicky, Adv. Synth. Catal. 2013, 355, 1869. doi: 10.1002/adsc.201300284. [2] (a) J. He, L. G. Hamann, H. M. L. Davies, R. E. J. Beckwith, Nat. Commun. 2015, 6, 5943. doi: 10.1038/ncomms6943.(b) R. A. Aycock, C. J. Pratt, N. T. Jui, ACS Catal. 2018, 8, 9115. doi: 10.1021/acscatal.8b03031.
(c) J. Chan, Y. Chang, M. Wasa, Org. Lett. 2019, 21, 984. doi: 10.1021/acs.orglett.8b03959.
(d) B. T. Matsuo, J. T. M. Correia, M. W. Paixão, Org. Lett. 2020, 22, 7891. doi: 10.1021/acs.orglett.0c02821.
(e) J. P. Barham, M. P. John, J. A. Murphy, J. Am. Chem. Soc. 2016, 138, 15482. doi: 10.1021/jacs.6b09690.
[3] (a) M. N. Hopkinson, B. Sahoo, J. L. Li, F. Glorius, Chem. – Eur. J. 2014, 20, 3874. doi: 10.1002/chem.201304823.(b) K. L. Skubi, T. R. Blum, T. P. Yoon, Chem. Rev. 2016, 116, 10035. doi: 10.1021/acs.chemrev.6b00018.
(c) M. D. Levin, S. Kim, F. D. Toste, ACS Cent. Sci. 2016, 2, 293. doi: 10.1021/acscentsci.6b00090.
[4] (a) G. Revol, T. McCallum, M. Morin, F. Gagosz, L. Barriault, Angew. Chem. Int. Ed. 2013, 52, 13342. doi: 10.1002/anie.201306727.(b) K. Shimomaki, K. Murata, R. Martin, N. V. Iwasawa, J. Am. Chem. Soc. 2017, 139, 9467. doi: 10.1021/jacs.7b04838.
(c) K. Murata, N. Numasawa, K. Shimomaki, J. Takaya, N. Iwasawa, Chem. Commun. 2017, 53, 3098. doi: 10.1039/C7CC00678K.
[5] C. Remeur, C. B. Kelly, N. R. Patel, G. A. Molander, ACS Catal. 2017, 7, 6065. doi: 10.1021/acscatal.7b01973. [6] (a) F. D. Lewis, T. I. Ho, J. Am. Chem. Soc. 1980, 102, 1751. doi: 10.1021/ja00525a061.(b) X. Zhang, S. R. Yeh, S. Hong, M. Freccero, A. Albini, D. E. Falvey, P. S. Mariano, J. Am. Chem. Soc. 1994, 116, 4211. doi: 10.1021/ja00089a010.
(c) L. Ruiz Espelt, I. S. McPherson, E. M. Wiensch, T. P. Yoon, J. Am. Chem. Soc. 2015, 137, 2452. doi: 10.1021/ja512746q.
[7] (a) S. M. Thullen, T. Rovis, J. Am. Chem. Soc. 2017, 139, 15504. doi: 10.1021/jacs.7b09252.(b) M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu, T. Rovis, Angew. Chem. Int. Ed. 2019, 58, 4002. doi: 10.1002/anie.201812227.
[8] (a) K. T. Sylvester, K. Wu, A. G. Doyle, J. Am. Chem. Soc. 2012, 134, 16967. doi: 10.1021/ja3079362.(b) C. Heinz, J. P. Lutz, E. M. Simmons, M. M. Miller, W. R. Ewing, A. G. Doyle, J. Am. Chem. Soc. 2018, 140, 2292. doi: 10.1021/jacs.7b12212.
[9] D. J. Sepelak, C. G. Pierpont, E. K. Barefield, J. T. Budz, C. A. Poffenberger, J. Am. Chem. Soc. 1976, 98, 6178. doi: 10.1021/ja00436a018.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载














No comments yet.