
本文作者alberto-caeiro
芝加哥大学董广彬教授开发了一种非高张力和高极性体系的C-C键活化新策略,为多取代苯酚的合成提供了新方法。
背景
过渡金属催化剂作为一种条件温和,选择性强的方法,在C-C键活化中变得更加重要。然而,现阶段的C-C键活化被局限在高张力或高度极化的体系中,非极性或者非张力的C-C键活化仍然是一大难题。
2018年,董广斌报道了一种在2,2′-biphenols(BINOL)中非张力的C(aryl)–C(aryl)键的活化。该方法的关键是利用羟基作为位点,将导向基配体磷酸酯引入体系,通过与金属Rh配位后活化C-C键,并用H2作为还原剂,得到苯酚衍生物。该方法具有低的催化剂负载量和好的官能团耐受性,并且可用于合成2,3,4-三取代苯酚。机理研究表明C-C键活化步骤是由Rh(l)-H物种催化的。
Catalytic activation of unstrained C(aryl)–C(aryl) bonds in 2,2′-biphenols
Zhu, Jun; Wang, Jianchun; and Dong, Guangbin*Nat. Chem.,2019, 45.DOI: s41557-018-0157-x.
工作介绍
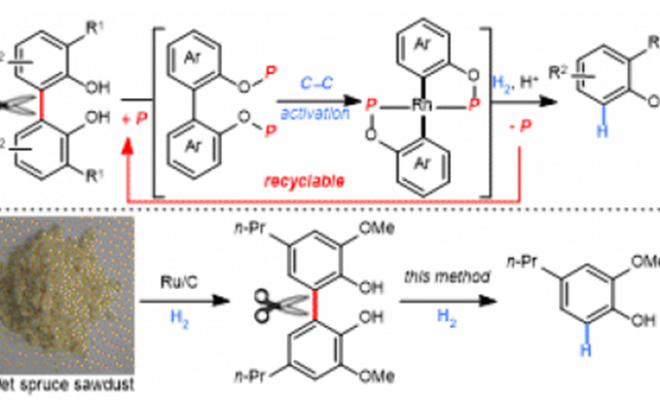
1994年,Milstein教授开创性的发现了通过Pincer类型的底物[1],实现了芳基烷基C-C键的直接氧化加成,该过程的驱动力是形成两个金属杂5元环(图1a)。虽然联芳烃化合物在现今有极大的运用,但这类非张力化合物的直接C-C键活化面临这3个难以解决的问题。1:与常用作C-C键活化的酮类底物不同,非极性的C-C键缺少可与富电子的金属反应的缺电子中心;2:联芳烃的C-C键与过渡金属d轨道的重叠会因为联芳烃扭转的构形而变得更加困难,例如,2,2’-而取代联苯酚,扭转角接近90°[2],从而难以形成最初的σ键配位物种(图1b);3:sp2(C)-sp2(C)的键解离能(>110kJ/mol)明显大于普通的C-C键的键解离能(如:C(acyl)–C(alkyl)为82 kJ/mol)。因此,反应的关键是如何控制联芳烃的构形,并将低价态的过渡金属尽可能的接近目标C-C键。作者在2,2′-联苯酚中通过将羟基作为handle,将可循环的导向基——磷酸酯键合进体系[3],从而导向金属至预期的C-C键处,实现C-C键活化反应(图1c)。

图1:a: Milstein最初C-C键切断反应; b: 2,2’-联苯酚扭转的构形; c: 2,2’-联苯酚的C-C键活化反应.
通过一系列的条件优化,作者得到了最佳条件(详见文章SI)。随后为了弄清各个反应物的作用,作者做了一系列控制实验(表1)。当没有金属时没有反应(entry2),更换磷酸酯配体时,效果会变差或不反应(entry3&4),作者推测缺电子的磷酸酯可能会发生C-P键的氧化加成,更换金属种类时,反应不发生(entry11&12),降低催化剂当量也不反应(entry8)。
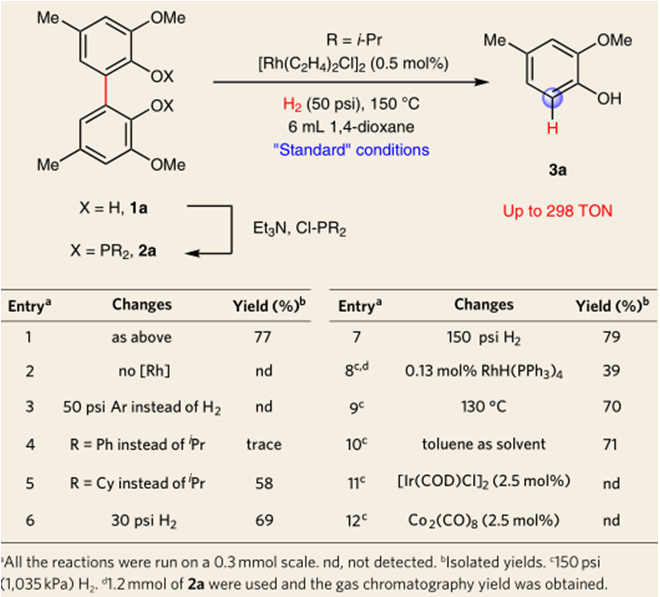
表1:条件筛选及控制实验
作者随后对底物进行了拓展,如表2。底物的4号位上有很好的普适性,各类烷基,芳基都可以很好的兼容,但当位阻偏大时,需要更多的催化剂和更高的压力;各种官能团也能兼容,氰基、酯基、酰胺基、烷基胺、烷基醚都能有中等到良好的收率,对于过渡金属催化剂有很强毒性的硫原子,反应也有中等收率;杂原子也能有效的兼容。
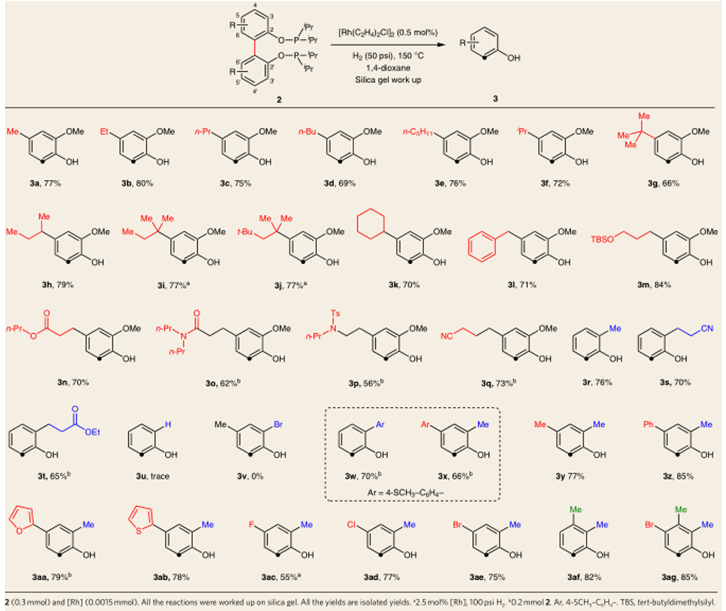
表2:底物范围
随后作者根据之前报道的文章,猜测可两种可行的机理(图2a)。Patha: Rh(l)-Cl直接对C-C键进行氧化加成,得到双芳基Rh(lll)物种,随后再与H2氧化加成,随后两次还原消除得到产物,因此,patha经过一个Rh(V)物种。Pathb: Rh(l)-Cl先于H2反应得到Rh(l)-H物种,它比Rh(l)-Cl更富电子,更有可能作为真实的催化剂,Rh(l)-H经过C-C键的氧化加成得到Rh(lll)物种,随后还原消除得到一个产物和芳基Rh(l)物种,随后与H2氧化加成,在还原消除得到另一分子产物和活性催化剂Rh(l)-H物种,因此,pathb只包含Rh(I) and Rh(lll)中间体。
为了区分这两种机理,作者做了机理计算(见文章SI)和控制实验。将从Ph2PCl得到的底物与[Rh(C2H4)2Cl]2反应得到含Cl桥键的复合物,而当没有H2存在时,同样反应只能得到衡量的产物。DFT计算表明,Rh(l)-Cl对C-C键的氧化加成是可逆的,并且随后对H2的氧化加成有很高的能垒(34.2kJ/mol)。但在pathb中,Rh(l)-Cl与H2的氧化加成会产生一分子的HCl,HCl会使P-O键断裂。实际上,当催化剂当量增加时,产率会降低,而当加入碱时,产物会恢复至原有水平(图2c)。这些表明反应中HCl是有生成的,反应中的键有两种作用,一是促进Rh(l)-H物种形成,二是中和生成的HCl防止P-O键断裂。当Rh(PPh3 ) 4H作为假设的催化剂与底物3c反应时,无需H2,加热可直接得到产物。这个结果强烈的表明pathb是更可行的机理。DFT机理也表明,pathb在能量上比patha更加有利。计算表明,反应决速步为C-C键断裂,能垒为28.1kJ/mol。如果Rh(l)-H可以活化C-C键,那么芳基或烷基Rh物种也可发生此类反应。于是作者将底物与Rh(l)-Cl加合物与PhLi混合发生转金属反应,过滤掉LiCl后,加热该复合物,如预期那样,得到了C-C偶联的产物(图2d)。这些证据都表明pathb为更可行的机理。
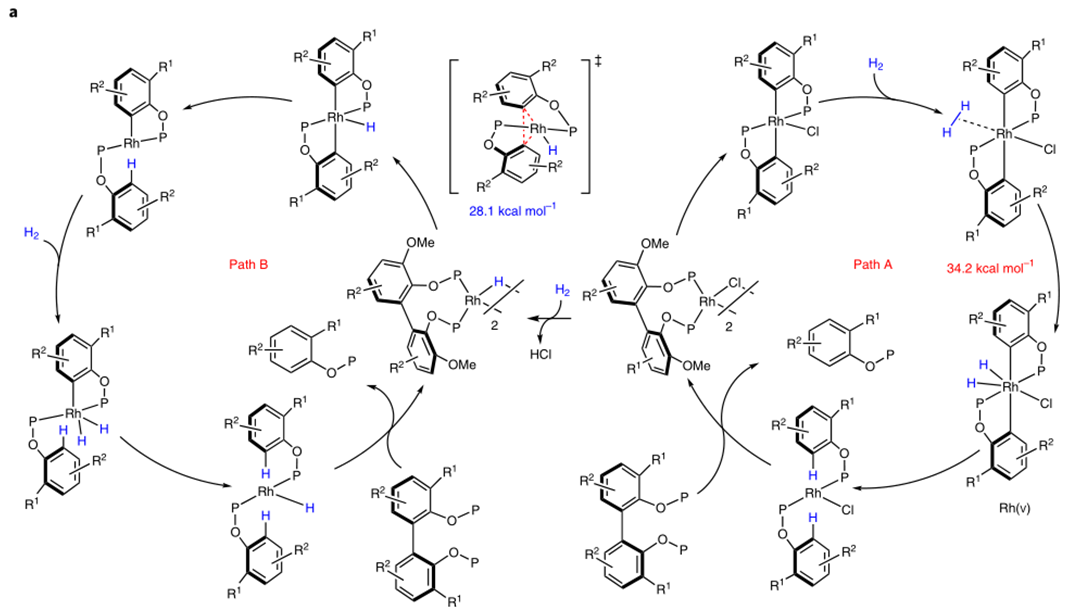

图2:机理研究。
作者对反应的应用性也做了研究。如图3a,反应可用于2,3,4-三取代苯酚的合成。图3b,反应可进行克级放大。图3c,一锅法也可以以中等收率得到产物。图3d,反应中的磷酸酯配体可以重复使用。图4中,作者对木质素的二聚体的C-C键断裂进行了研究。
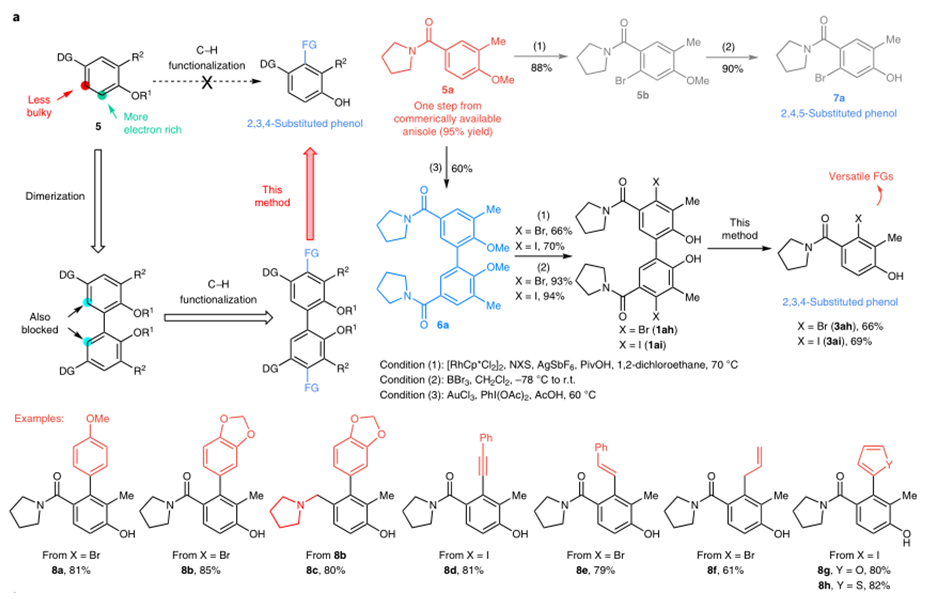
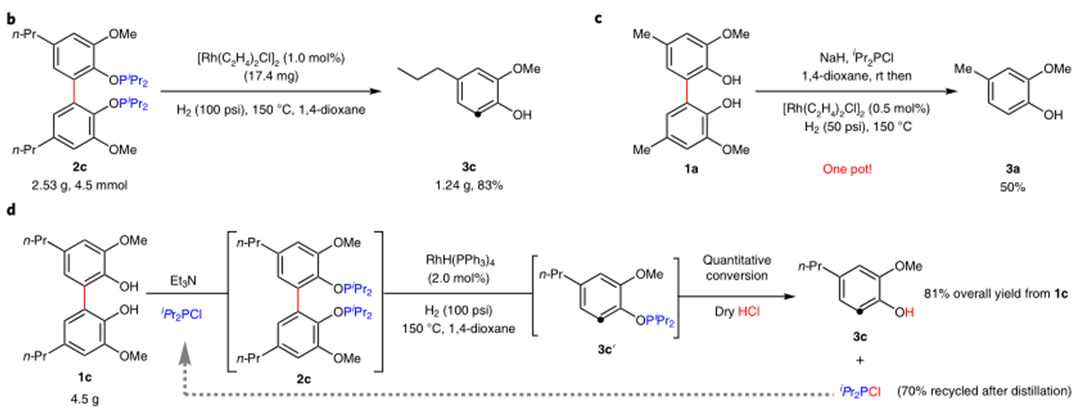
图3:Catalytic reductive cleavage of C(aryl)-C(aryl) bonds in 2,2′-biphenols
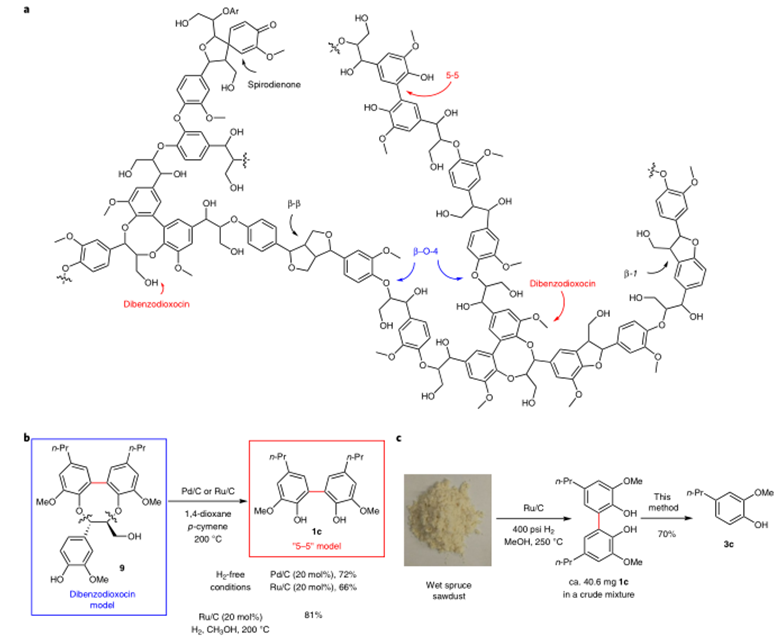
图4:model study for the cleavage of C-C bonds in lignin dimers
参考文献
[1]: a) Gozin, M., Weisman, A., Ben-David, Y., Milstein, D. Nature 1993, 364,699. doi.org/10.1038/364699a0. b) Gozin, M. Milstein, D.Nature1994, 370, 42. doi.org/10.1038/370042a0. [2]: Grein, F. J. Phys. Chem. A, 2002, 106, 3823.DOI: 10.1021/jp0122124. [3]: Rousseau, G.,Breit, B. Angew. Chem. Int. Ed., 2011, 50, 2450.doi.org/10.1002/anie.201006139.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!

















No comments yet.