
本文作者:杉杉
导读
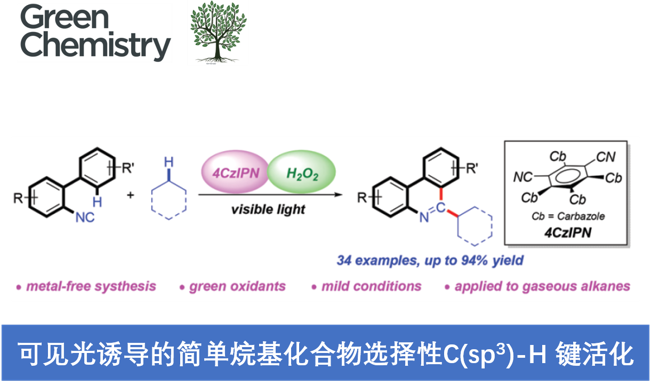
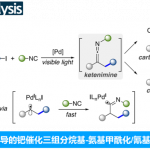
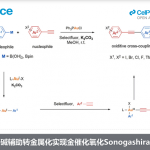
近日,大连理工大学的段春迎与金云鹤课题组在Green Chemistry中发表论文,报道一种采用可见光诱导的无金属条件下的简单烷基化合物与异腈之间的C(sp3)-H键菲啶化 (phenanthridinylation)反应方法学。其中,选择H2O2作为终端氧化剂。同时,这一全新的菲啶化策略具有反应条件温和、底物应用范围广泛以及良好的选择性与反应收率等优势。值得注意的是,各类气态烷烃同样能够有效地参与上述的菲啶化过程,并获得一系列具有良好应用价值的烷基取代菲啶 (alkyl-substituted phenanthridine)衍生物。
Selective C(sp3)-H activation of simple alkanes: visible light-induced metal-free synthesis of phenanthridines with H2O2 as a sustainable oxidant
Y. Zhang, Y. Jin, L. Wang, Q. Zhang, C. Meng, C. Duan, Green Chem. ASAP. doi:10.1039/d1gc02670d.
正文
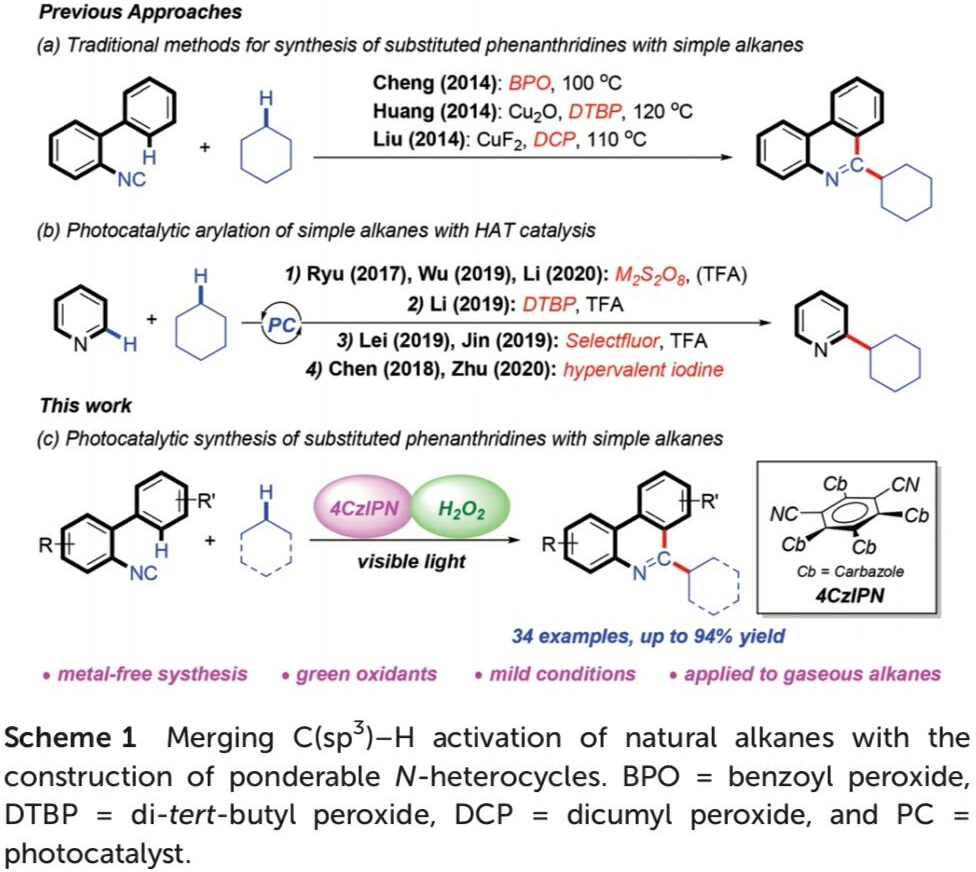
N-杂环化合物广泛存在于各类天然产物以及生物活性分子中。同时,在新药研发过程中发挥较为重要的作用。尤其菲啶作为典型的N-杂环化合物,已经广泛应用于医药化学的相关研究,并表现出不同的生物活性,例如抗菌、抗肿瘤、细胞毒性以及抗白血病活性。在过去的十年中,选择2-异氰基联芳底物进行的自由基加成串联反应方法学,已经成为合成6-取代菲啶衍生物的一种较为实用的反应策略[1]。另一方面,简单烷基化合物,作为石油与天然气中的主要组分,广泛存在于自然界中。然而,由于其固有的分子惰性以及反应过程中化学选择性的不可调控性,因此,实现简单烷基化合物中C(sp3)-H键的直接活化,具有极高的挑战性[2]。并且,将各类烷基化合物应用于一系列重要N-杂环化合物的构建,在合成化学中同样面临巨大的挑战[3]-[6]。2014年,Cheng[4]、Huang[5]以及Liu[6]课题组分别报道采用有机过氧化物促进的简单烷基化合物与异腈之间的菲啶基化反应策略,并且,反应过程中均涉及自由基中间体 (Scheme 1a)。
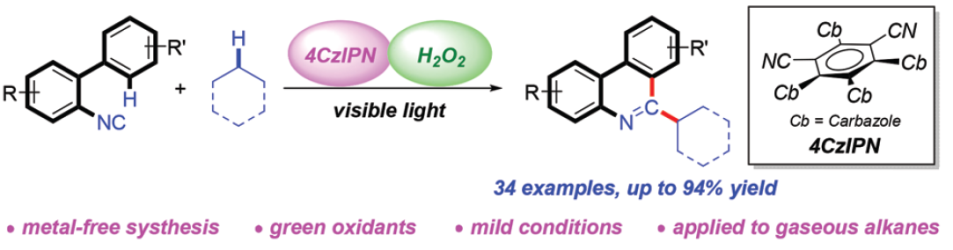
近年来,可见光诱导的光氧化还原氢原子转移 (HAT)催化方法学,已经发展成为实现简单烷基化合物官能团化的一种有效策略[7]。通过这一策略,诸多课题组成功实现各类简单烷基化合物与N-杂环化合物之间的芳基化反应[8],然而,在上述策略中,仍需要采用过量的氧化剂,例如过硫酸盐、DTBP、Selectfluor以及超共价碘试剂,进而产生大量废弃的还原副产物 (Scheme 1b)。为避免上述问题,作者选择H2O2水溶液作为绿色氧化剂,并将其应用于N-杂芳基化合物与脂肪族C-H键之间的脱氢交叉偶联反应方法学的研究。基于上述的文献报道以及本课题组长期以来对于采用可见光促进的相关合成转化策略的研究。这里,本文将报道一种光催化的HAT反应策略,通过2-异氰基联芳与简单烷基化合物作为起始原料,进而在无金属试剂存在的条件下,成功实现一系列烷基取代菲啶类化合物的合成 (Scheme 1c)。其中,4-CzIPN作为光催化剂,绿色氧化剂H2O2作为具有HAT-活性的羟基自由基前体。
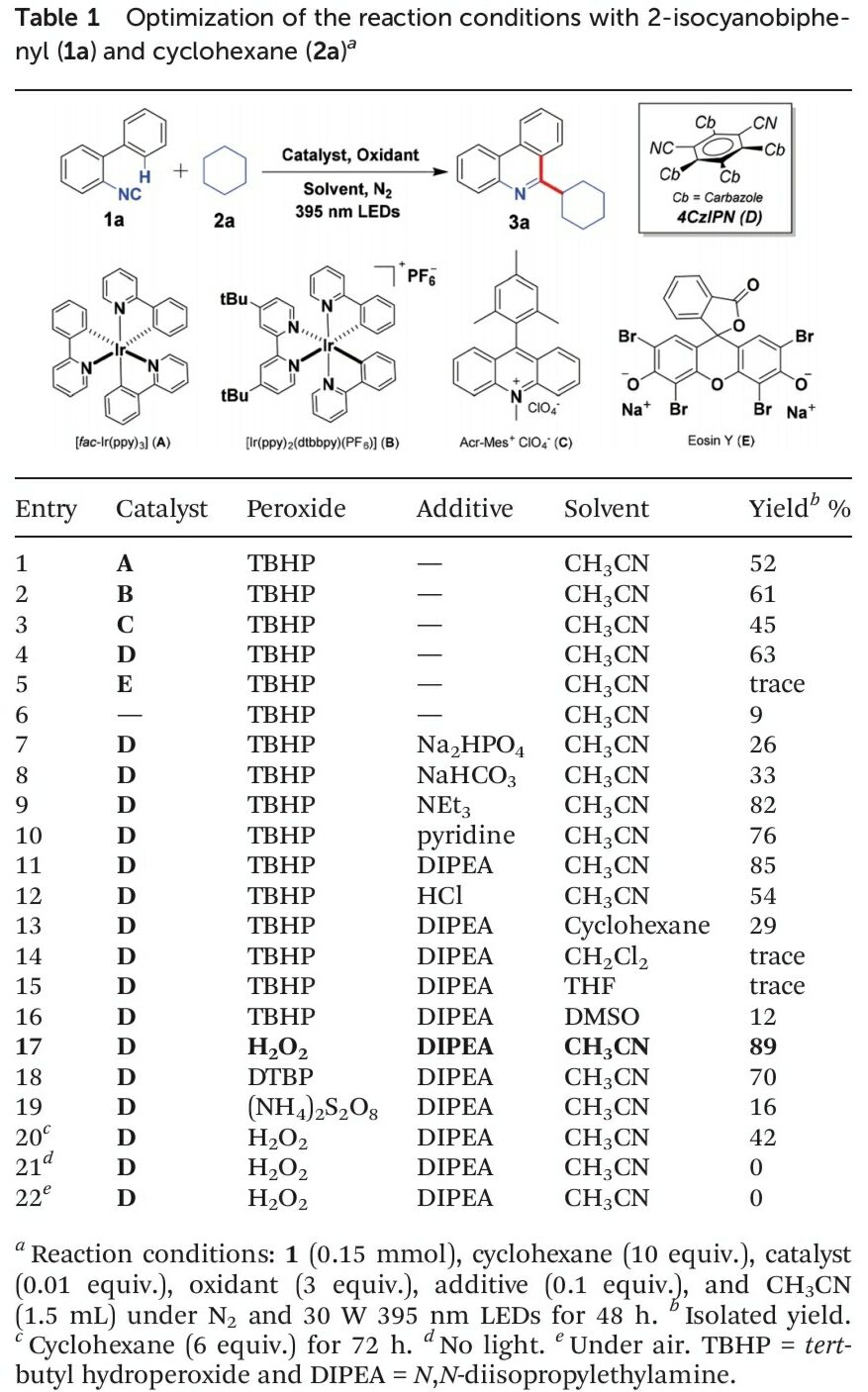
首先,作者采用2-异氰基-1,1′-联苯1a与环己烷2a作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用4-CzIPN作为光催化剂,H2O2作为终端氧化剂,DIPEA作为添加剂,在乙腈溶剂以及30 W 395 nm LED辐射条件下进行反应,最终获得89%收率的6-环己基菲啶产物3a。
在上述的最佳反应条件下,作者首先对2-异氰基联芳底物的适用范围进行考察 (Table 2, left column)。研究表明,芳环中具有供电子与吸电子基团取代的2-异氰基联芳底物,均能较好地与上述的标准反应条件兼容,并获得相应的目标产物3a–3p,收率为58-90%。值得注意的是,通过这一全新的菲啶化策略,能够成功实现重要生物碱,即烷基取代的trisphaeridine (3p)分子的构建。同时,为进一步阐明上述菲啶化策略的合成应用价值,作者进一步进行1a (5.6 mmol, 1.00 g)与2a 之间菲啶化过程的克级规模实验研究。最终,作者发现,产物3a的收率 (1.24 g, 84.8% 反应收率)无显著降低。
接下来,该小组进一步对简单烷基底物的应用范围进行考察 (Table 2, right column)。研究表明,一系列环状与线性的烷基底物,在上述的标准反应条件下,均能够顺利地与2-异氰基联苯进行反应,并获得相应烷基取代的菲啶产物3q–3aa,收率为45-87%。同时,该小组同样对烷基底物在反应过程中,不同位置的区域异构比 (regiomeric ratio, r.r.)进行研究。实验结果 (例如3w产物中,r.r.为1.8 : 1,而3x 产物中,r.r.为1 : 1.8)表明,反应过程的区域选择性受到相应碳自由基的稳定性以及相应氢原子化学环境的控制[9]-[10]。同时,在上述的标准反应体系中,并未观察到相应一级碳自由基捕获的产物。接下来,作者发现,液化石油气 (liquid petroleum gas, LPG)中的重要组分正丁烷与异丁烷,同样能够有效地参与上述的菲啶化过程,并以中等程度的反应收率,获得相应的目标产物3z与3aa。此外,该小组进一步发现,醚类、胺类、醛以及甲酰胺底物中的C(sp2)-H键同样能够有效地参与上述的菲啶化过程,并以良好的区域选择性(对于醚以及胺类底物,反应优先在相应杂原子的α-位置进行,而对于醛类及甲酰胺底物,则优先进行C(sp2)-H键的活化)与中等至良好的反应收率,获得相应产物3ab–3ah。
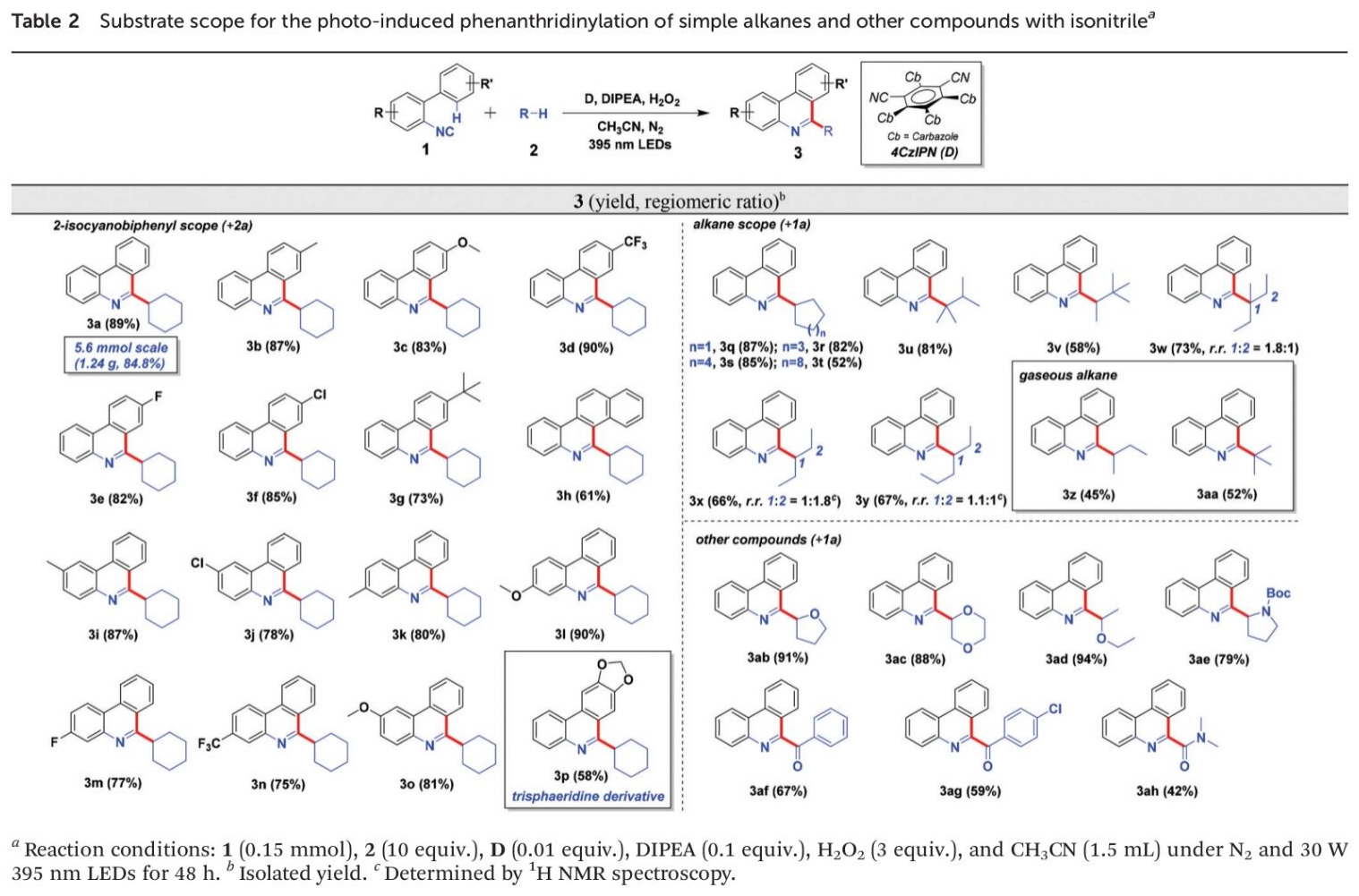
接下来,为提出合理的反应机理,作者进行一系列相关的实验研究 (Scheme 2)。首先,该小组进行自由基捕获实验 (Scheme 2a)的研究。在实验过程中,作者观察到,在1a与2a的标准反应体系中加入自由基捕获剂TEMPO时,反应受到完全地抑制,并通过GC-MS以及HRMS检测出TEMPO与环己基自由基之间形成的相应加合物,进而表明反应过程中涉及自由基中间体 (Scheme 2a)。之后,作者通过 (a) 分别采用1a以及d5-1a底物,在上述的标准反应条件下形成产物d4–3a (b)采用1a与d5-1a 1 : 1的混合物,在上述的标准反应条件下形成产物d4–3a 的两种方式,对反应反应过程中C(sp2)-H键断裂的KIE进行研究。并分别通过kH/kD[11]以及PH/PD[11],测定出C(sp2)-H键断裂时的KIE数值分别为1.0与1.5。同时,作者同样通过环己烷与d12–环己烷作为反应底物,并同样采用kH/kD以及PH/PD的方式,测定出C(sp3)-H键断裂时的KIE数值为1.9与3.0 (Scheme 2b)。上述的KIE数值表明,通过自由基中间体进行的HAT过程为决速步骤,而C(sp2)-H键断裂的过程则为上述催化反应过程中的产物决定步骤 (Scheme 2b)。此外,该小组通过Stern-Volmer荧光淬灭实验 (Stern-Volmer fluorescence quenching experiment),对反应过程中的相关SET过程进行研究 (Scheme 2c)。作者发现,4-CzIPN在405 nm处激发时,能够在548 nm处观察到相应的荧光。在进一步加入DIPEA时,能够使荧光强度显著降低。同时,作者通过后续的实验研究表明,反应体系中存在的其它试剂 (例如H2O2与环己烷),则无法产生相应的荧光淬灭 (详见SI)。因此,通过Stern-Volmer荧光淬灭实验能够表明,4-CzIPN与DIPEA之间存在SET过程。接下来,作者发现,在形成产物3a时,反应过程的量子产率为0.033,进而表明自由基链反应过程对于目标产物的形成无显著贡献[12]。
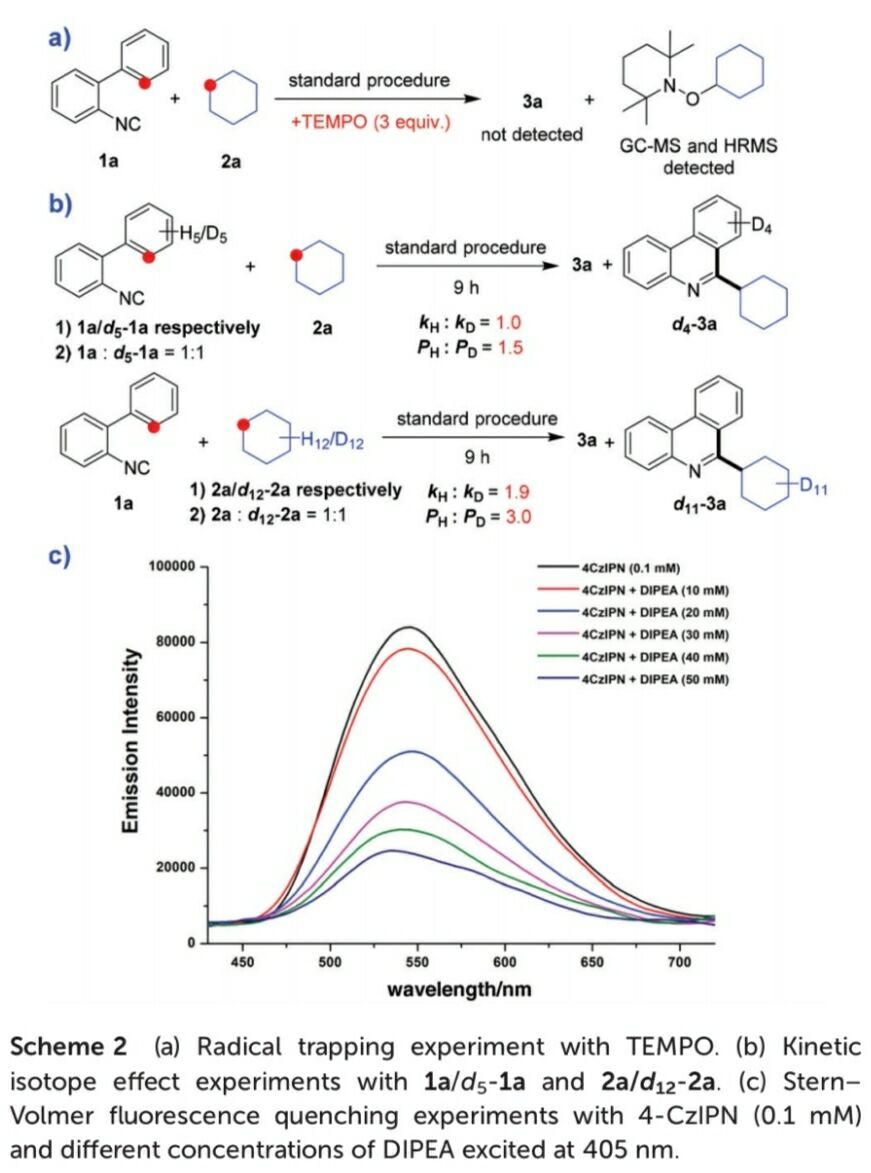
基于上述的实验研究,作者提出一种合理的反应机理路径 (Scheme 3)。首先,在光辐射条件下,光催化剂4-CzIPN转化为激发态I。之后,激发态I与DIPEA通过SET过程,生成自由基II与III。同时,自由基II与H2O2再次通过SET过程,形成羟基负离子以及具有HAT-活性的羟基自由基,并伴随光催化剂4-CzIPN的再生。接下来,通过烷基化合物与羟基自由基之间的HAT过程,形成烷基自由基IV。并进一步通过IV与2a之间的自由基加成过程,形成亚胺基自由基 (imidoyl radical)V,V继续经历分子内芳基加成过程,形成自由基中间体VI。最后,中间体VI、自由基III以及羟基负离子之间,经历进一步的氧化脱氢 (oxidative dehydrogenation)过程,获得最终的目标产物3a。同时,使DIPEA再生。此外,研究发现,在反应体系中无DIPEA存在时,通过激发态光敏剂I与H2O2之间进行的能量转移 (ENT)过程,同样能够进一步获得具有HAT-活性的羟基自由基,然而,却无法进行相应的SET过程 (path B)。
总结
大连理工大学段春迎与金云鹤课题组报道一种温和与绿色的催化体系,采用H2O2作为终端氧化剂,在可见光诱导以及无金属试剂存在的条件下,通过简单烷基化合物的C(sp3)-H键活化策略,成功完成一系列取代菲啶衍生物的合成。这一全新的菲啶化策略具有反应条件温和、环境友好、底物应用范围广泛、能够适用于气态烷烃、良好的官能团兼容性以及原料成本低廉等优势。此外,上述策略同样能够成功用于天然生物碱烷基取代trisphaeridine的合成。
参考文献
[1] (a) M. Tobisu, K. Koh, T. Furukawa, N. Chatani, Angew. Chem. Int. Ed. 2012, 51, 11363. doi: 10.1002/anie.201206115.(b) H. Jiang, Y. Cheng, R. Wang, M. Zheng, Y. Zhang, S. Yu, Angew. Chem. Int. Ed. 2013, 52, 13289. doi: 10.1002/anie.201308376.
(c) B. Zhang, C. G. Daniliuc, A. Studer, Org. Lett. 2014, 16, 250. doi: 10.1021/ol403256e.
(d) J. Liu, C. Fan, H. Yin, C. Qin, G. Zhang, X. Zhang, H. Yi, A. Lei, Chem. Commun. 2014, 50, 2145. doi: 10.1039/C3CC49026B.
(e) T. Xiao, L. Li, G. Lin, Q. Wang, P. Zhang, Z. Mao, L. Zhou, Green Chem. 2014, 16, 2418. doi: 10.1039/C3GC42517G.
[2] N. Gunsalus, J. A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi, R. A. Periana, Chem. Rev. 2017, 117, 8521. doi: 10.1021/acs.chemrev.6b00739. [3] (a) A. P. Antonchick, L. Burgmann, Angew. Chem. Int. Ed. 2013, 52, 3267. doi: 10.1002/anie.201209584.(b) L. Zhou, H. Togo, Eur. J. Org. Chem. 2019, 7, 1627. doi: 10.1002/ejoc.201801797.
[4] W. Sha, J. Yu, Y. Jiang, H. Yang, J. Cheng, Chem. Commun. 2014, 50, 9179. doi: 10.1039/C4CC03304C. [5] Z. Zhu, T. Wang, P. Bai, Z. Huang, Org. Biomol. Chem. 2014, 12, 5839. doi: 10.1039/C4OB01256A. [6] Z. Li, F. Fan, J. Yang, Z. Liu, Org. Lett. 2014, 16, 3396. doi: 10.1021/ol501461u. [7] (a) A. Hu, J. Guo, H. Pan, Z. Zuo, Science 2018, 361, 668. doi: 10.1126/science.aat9750.(b) H. Deng, Q. Zhou, J. Wu, Angew. Chem. Int. Ed. 2018, 57, 12661. doi: 10.1002/anie.201804844.
(c) G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun, T. Noël, Science 2020, 369, 92. doi: 10.1126/science.abb4688.
(d) S. Rohe, A. O. Morris, T. McCallum, L. Barriault, Angew. Chem. Int. Ed. 2018, 57, 15664. doi: 10.1002/anie.201810187.
(e) B. J. Shields, A. G. Doyle, J. Am. Chem. Soc. 2016, 138, 12719. doi: 10.1021/jacs.6b08397.
(f) S. M. Treacy, T. Rovis, J. Am. Chem. Soc. 2021, 143, 2729. doi: 10.1021/jacs.1c00687.
(g) D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa, I. Ryu, ACS Catal. 2018, 8, 701. doi: 10.1021/acscatal.7b03354.
[8] (a) C. Huang, J. Wang, J. Qiao, X. Fan, B. Chen, C. Tung, L. Wu, J. Org. Chem. 2019, 84, 12904. doi: 10.1021/acs.joc.9b01603.(b) H. Tian, H. Yang, C. Tian, G. An, G. Li, Org. Lett. 2020, 22, 7709. doi: 10.1021/acs.orglett.0c02912.
(c) X. A. Liang, L. Niu, S. Wang, J. Liu, A. Lei, Org. Lett. 2019, 21, 2441. doi: 10.1021/acs.orglett.9b00744.
(d) H. Zhao, J. Jin, Org. Lett. 2019, 21, 6179. doi: 10.1021/acs.orglett.9b01635.
(e) X. Shao, X. Wu, S. Wu, C. Zhu, Org. Lett. 2020, 22, 7450. doi: 10.1021/acs.orglett.0c02475.
[9] Y. Jin, Q. Zhang, L. Wang, X. Wang, C. Meng, C. Duan, Green Chem. 2021, ASAP. doi: 10.1039/d1gc01563j. [10] Q. An, Z. Wang, Y. Chen, X. Wang, K. Zhang, H. Pan, W. Liu, Z. Zuo, J. Am. Chem. Soc. 2020, 142, 6216. doi: 10.1021/jacs.0c00212. [11] E. M. Simmons, J. F. Hartwig, Angew. Chem. Int. Ed. 2012, 51, 3066. doi: 10.1002/anie.201107334. [12] M. A. Cismesia, T. P. Yoon, Chem. Sci. 2015, 6, 5426. doi: 10.1039/C5SC02185E.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!















No comments yet.