
本文作者:杉杉
导读
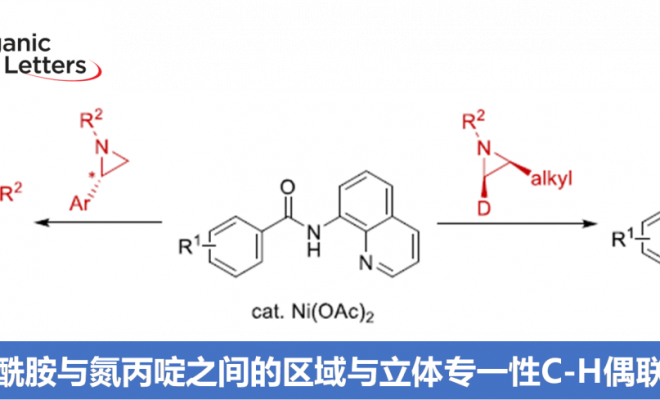
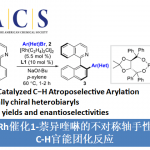
本文主要报道在镍催化条件下,8-氨基喹啉衍生的苯甲酰胺与芳基以及烷基取代氮丙啶之间的C-H偶联反应方法学。这一策略中,涉及串联C-H烷基化/分子内酰胺化过程,同时,伴随氨基喹啉辅基的去除。此外,氮丙啶开环过程的区域选择性能够通过底物中取代基的不同进行控制。同时,实验过程中作者观察到,手性氮丙啶的立体构型发生翻转,进而表明氮丙啶开环的机理涉及SN2型亲核开环步骤。其中,日本Osaka大学Hirano Koji (平野 康次)与Miura Masahiro (三浦 雅博)为共同通讯作者。
Nickel-Catalyzed Regio- and Stereospecific C−H Coupling of Benzamides with Aziridines
S.Xu, K.Hirano, M. Miura, Lett.2021, 23, 5471. doi: 10.1021/acs.orglett.1c01821.
正文
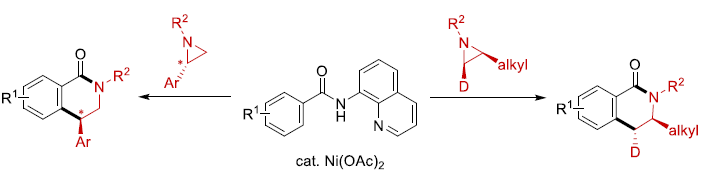
氮丙啶化合物由于其固有的环张力,已成为构建结构较为复杂的含氮有机分子的重要砌块。其中, 通过Lewis酸促进的由富电子芳环参与的Friedel-Crafts烷基化反应方法学以及金属催化的采用芳卤或有机金属试剂参与的交叉偶联反应方法学,在构建C-C键方面已取得诸多的研究进展。尤其是通过金属催化的芳烃与氮丙啶之间的C-H偶联过程,构建β-芳基乙胺的反应策略[1],由于具有良好的步骤与原子经济性,因而备受广泛关注。Li课题组[2] 开创性地报道了采用Cp*Rh(III)催化的2-芳基吡啶 (其中吡啶作为导向基团)与氮丙啶之间的邻位C-H烷基化反应方法学。之后,Yoshikai (吉戒 直彦)课题组[3]研究发现,采用Co-NHC催化体系,同样能够有效地实现上述吡啶导向的邻位C-H烷基化过程 (Scheme 1a, top)。然而,在上述的反应策略中,底物范围仅局限于反应活性较高的芳基取代氮丙啶,而对于烷基取代氮丙啶底物的C-H偶联反应,则较少有文献报道。目前仅有一例成功的文献报道,即Zhao课题组[4]报道的采用钯催化剂促进的苯甲酸与烷基取代氮丙啶之间通过羧酸辅助的邻位C-C偶联反应方法学 (carboxylic-acid-assisted ortho-C−C coupling, Scheme 1a, bottom)。与此同时,本课题组近期已经报道镍催化的苯甲酰胺与环氧化物或氧杂环丁烷之间的C-H偶联反应方法学[5]。其中,关键步骤涉及N,N-双齿氨基喹啉辅基 (N,N-bidentate aminoquinoline auxiliary)的辅助,并且,通过氨基喹啉导向基的移除,最终获得相应的六元环与七元环benzolactone产物。尤其值得关注的是,在采用内环氧化物 (internal epoxide)参与的C-C键形成过程中,观察到立体构型的保持。受到上述研究的启发,这里,本文报道采用镍催化剂促进的8-氨基喹啉衍生的苯甲酰胺与芳基或烷基取代氮丙啶之间的C-H偶联反应策略 (Scheme 1b)。这一策略中,首先通过N,N-双齿螯合促进相应C-H的烷基化,接下来,通过分子内酰胺化过程,进而形成相应的六元环内酰胺衍生物,同时,反应过程中伴随氨基喹啉导向基团的去除。同时,研究发现,2-芳基以及2-烷基取代的氮丙啶底物均能够顺利地参与上述反应,最终获得官能团化的3,4-二氢异喹啉酮 (3,4-dihydroisoquinolinone)产物。值得注意的是,上述镍催化的反应过程具有高度的立体专一性。并且,反应过程中,手性氮丙啶的立体构型发生翻转,进而表明氮丙啶的开环机理涉及氧化还原中性的SN2型开环过程,这与通过环氧化物参与的C-H偶联过程相反。
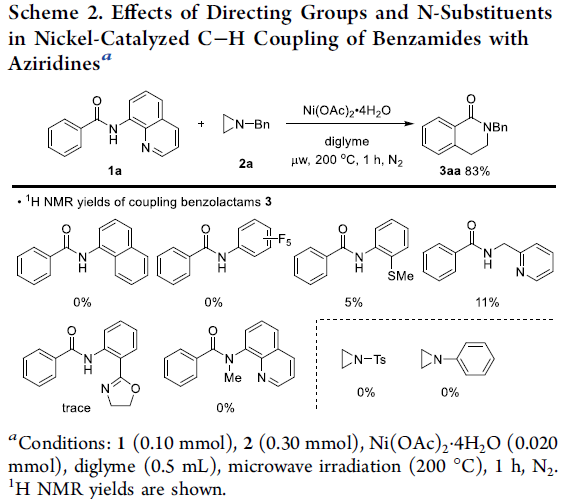
首先,作者采用苯甲酰胺衍生物1a与N-苄基氮丙啶2a作为模型底物,进行了相关偶联反应条件的优化筛选 (Scheme 2)。确定最佳的反应条件为:采用Ni(OAc)2作为催化剂,二甘醇二甲醚 (diglyme)作为反应溶剂,在微波辐射 (200 oC)条件下,反应1 h,即可获得83%收率的苯并内酰胺 (benzolactam)产物3aa。
在获得上述最佳反应条件后,作者开始对苯甲酰胺衍生物1的底物应用范围进行考察 (Scheme 3)。研究表明,芳基不同位置带有各类供电子基与吸电子基取代的苯甲酰胺衍生物,均能够与2a顺利地反应,获得相应的产物3aa-3la,收率为32-89%。同时,萘衍生物1m与1n同样能够有效地与2a进行相应的偶联过程,最终获得产物3ma与3na,表现出不同的区域选择性。此外,一系列噻吩衍生的酰胺底物,同样能够较好地适用于上述的偶联过程,进而成功实现一系列噻吩稠合内酰胺分子3oa–3pa的构建。值得注意的是,氨基喹啉导向基团能够自发地去除,并能够有效地进行回收。例如,在3aa的克级合成实验中,8-氨基喹啉的回收率为79%。

接下来,作者对氮丙啶底物2的应用范围进行深入研究 (Scheme 4)。该小组发现,在上述的标准反应条件下,2-烷基取代的氮丙啶均能够顺利地与1a进行相应的C-C偶联过程,并获得3-取代的3,4-二氢异喹啉酮产物3ab–3ad。同时,研究表明,采用光学纯的氮丙啶底物(S)-2e时,同样能够与1a进一步反应,形成手性产物(S)-3ae,同时,并未观察到产物对映纯度的丧失。

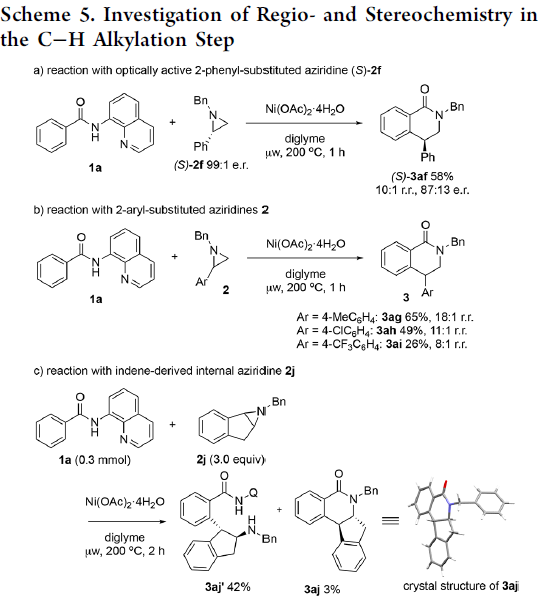
为提出合理的反应机理,作者进行一系列相关的实验研究 (Scheme 5)。首先,作者发现,将1a与对映纯的(S)-2f在标准条件下反应时,优先经历苄基C-N断裂,获得立体构型翻转的产物(S)-3af,并且,产物的对映体纯度略有降低 (Scheme 5a)。这一结果表明,镍能够促进苄基位置的正电荷的累积,使C-N键增长,并发生断裂。然而,反应机理步骤中并不涉及碳正离子中间体的参与。观察到的产物构型翻转,表明C-C偶联过程主要通过氧化还原中性的SN2型亲核开环途径进行。同时,作者进一步发现,采用芳基中带有吸电子与供电子基团取代的2-芳基吖丙啶底物2在上述标准条件下参与反应时,均观察到相同的区域选择性,并优先获得4-芳基二氢异喹啉酮产物3ag–3ai (Scheme 5b)。接下来,该小组发现,采用1a与茚衍生的氮丙啶底物2j在标准条件下进行反应时,同样优先经历苄基C-N断裂,并获得C-H烷基化产物3aj’ (主要产物)以及少量内酰胺产物3aj (Scheme 5c)。为深入研究反应过程的立体化学,作者将1a分别与cis–2k–d1以及trans–2k–d1在标准条件下反应,最终发现,反应过程具有较为理想的立体专一性,cis–2k–d1能够区域专一性地转化为trans–3ak–d1,而trans–2k–d1则区域专一性地转化为cis–3ak–d1,进而表明,镍催化过程具有高度的立体专一性。同时,其立体构型翻转的结果与环氧化物参与的相应C-H偶联反应相反。上述事实表明,镍催化剂在小环杂环底物的开环反应中,能够表现出较为独特的化学反应特性 (Scheme 5d)。
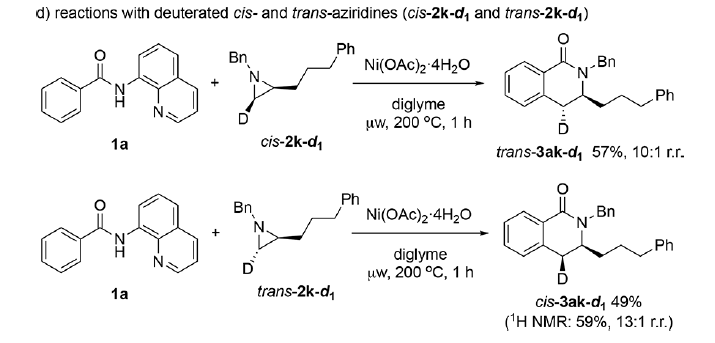
基于上述实验事实以及前期的文献报道,作者提出了一种合理的反应机理 (Scheme 6)。首先,通过苯甲酰胺衍生物 1a与Ni(OAc)2的螯合过程,形成Ni(II)配合物 A,配合物 A经历可逆的C-H断裂过程,形成环镍 (nickelacycle)中间体 B与HOAc。接下来,通过氮丙啶衍生物 2a中氮原子与环镍中间体 B中Ni中心的配位过程,使C-N键增长,并进一步促进C-C偶联过程的进行,通过SN2型亲核开环过程,形成中间体 D。根据反应过程中观察到的区域与立体化学的实验结果,能够排除涉及碳正离子的SN1型机理以及通过SET途径进行的吖丙啶开环机理。并且,反应过程中,环氧化物与吖丙啶底物开环机理的差异主要源自于氮原子与氧原子之间的Lewis碱性差异。其中,氮原子与金属中心具有相对较强的配位性能,能够进一步促进C-N键的极化,并利于SN2型亲核开环过程的进行。然而,环氧化物的开环过程则优先通过氧化还原活性的开环机理路径 (redox-active ring-opening pathway)[5a] 进行。同时,在 1a与对位取代芳基吖丙啶底物之间的反应中,作者通过Hammett图 (Hammett plot)观察到负斜率 (ρ =−1.09),进而表明决速步骤中涉及C-C键的形成过程 (参阅SI)。最后,中间体 D经历分子内酰胺化以及HOAc参与的质子分解 (protonolysis)过程,获得最终产物 3aa, 并回收8-氨基喹啉,同时,使Ni(OAc)2催化剂再生,进而完成相关的催化循环。
总结
本文主要报道通过镍催化促进的具有8-氨基喹啉导向基团的苯甲酰胺衍生物与一系列氮丙啶底物之间的C-H偶联反应方法学。其中,反应过程涉及C-H烷基化-分子内酰胺化的串联过程,进而成功实现一系列官能团化的3,4-二氢异喹啉酮分子的构建。值得注意的是,反应过程的区域选择性能够通过氮丙啶底物中取代基的不同进行控制。机理研究实验中观察到,上述的镍催化过程具有立体专一性,并且,C-C键的形成过程中涉及立体构型的翻转,进而表明反应过程涉及SN2型亲核开环途径。
参考文献
[1] (a) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes, M. Schnürch, Chem. Soc. Rev. 2018, 47, 6603. doi: 10.1039/C8CS00201K.(b) S. Rej, Y. Ano, N. Chatani, Chem. Rev. 2020, 120, 1788. doi: 10.1021/acs.chemrev.9b00495.
[2] X. Li, S. Yu, F. Wang, B. Wan, X. Yu, Angew. Chem. Int. Ed. 2013, 52, 2577. doi: 10.1002/anie.201209887. [3] (a) K. Gao, R. Paira, N. Yoshikai, Adv. Synth. Catal. 2014, 356, 1486. doi: 10.1002/adsc.201400049.(b) P. De, S. Atta, S. Pradhan, S. Banerjee, T. A. Shah, T. Punniyamurthy, J. Org. Chem. 2020, 85, 4785. doi: 10.1021/acs.joc.0c00010.
[4] K. Zhou, Y. Zhu, W. Fan, Y. Chen, X. Xu, J. Zhang, Y. Zhao, ACS Catal. 2019, 9, 6738. doi: 10.1021/acscatal.9b01347. [5] (a) S. Xu, K. Takamatsu, K. Hirano, M. Miura, Angew. Chem. Int. Ed. 2018, 57, 11797. doi: 10.1002/anie.201807664.(b) S. Xu, K. Takamatsu, K. Hirano, M. Miura, Chem. – Eur. J. 2019, 25, 9400. doi: 10.1002/chem.201900543.
















No comments yet.