

本文作者:Summer
导读
南开大学李鑫教授团队成功开发出采用B(C6F5)3/CPA (chiralphosphoric acid)催化体系促进的酮亚胺与非活化二烯之间的不对称氮杂Diels-Alder反应方法学,并以优秀的收率与对映选择性获得一系列含有季碳手性中心的四氢吡啶化合物。此外,作者通过DFT计算表明反应过程中立体选择性的控制源自于次级轨道相互作用 (secondary orbital interaction, SOI)与非共价相互作用 (non-covalent interaction, NCI)。
Catalytic Asymmetric Aza–Diels–Alder Reaction of Ketimines and UnactivatedDienes
Q. Zhao, Y. Li, Q. Zhang, J. Cheng, X. Li, Angew. Chem. Int. Ed. Early view. doi: 10.1002/anie.202104788.
正文
不对称氮杂Diels-Alder反应 (asymmetric aza-Diels-Alder reaction)可以有效地构建复杂结构的手性化合物,因而备受化学家的广泛关注。目前,在不对称氮杂Diels-Alder反应方法学的研究报道中,通常选择Danishefsky二烯、Brassard二烯以及环戊二烯等富电子的二烯,而对于简单的非活化二烯,例如,异戊二烯,在不对称氮杂Diels-Alder反应方法学中的应用则较少有文献报道[1]。此外,在上述方法学研究中所选择的亲二烯体通常为醛亚胺[2],而对于酮亚胺的应用同样较少有文献报道[3],可能源于其较低的反应活性与更加显著的立体位阻 (Figure 1A)。因此,发展非活化二烯与酮亚胺之间的不对称氮杂Diels-Alder反应方法学 (Figure 1B)面临巨大挑战,而解决这一问题的关键在于降低二者HOMO与LUMO之间的能隙。近期,南开大学的李鑫团队采用B(C6F5)3/CPA催化体系[4],成功实现由酮亚胺参与的不对称ene反应,即酮亚胺-ene反应 (ketimine-ene reaction)[5]。其中,B(C6F5)3能够显著提高CPA的酸性,从而降低反应的活化能垒。由此,作者设想通过选择B(C6F5)3/CPA催化剂体系,同样能够显著降低酮亚胺LUMO轨道的能量,使其能够与非活化二烯的HOMO进行良好地匹配,从而有效地实现酮亚胺与非活化二烯之间的不对称氮杂Diels-Alder反应。基于上述设想以及作者对一系列酮亚胺与酮亚胺-催化剂配合物亲电指数 (electrophilicity index)的理论计算,南开大学李鑫团队开发出采用B(C6F5)3/CPA催化的2-芳基-3H-吲哚-3-酮与非活化二烯之间的不对称氮杂Diels-Alder反应,并以优秀的收率与对映选择性获得一系列手性四氢吡啶化合物 (Figure 1C)。
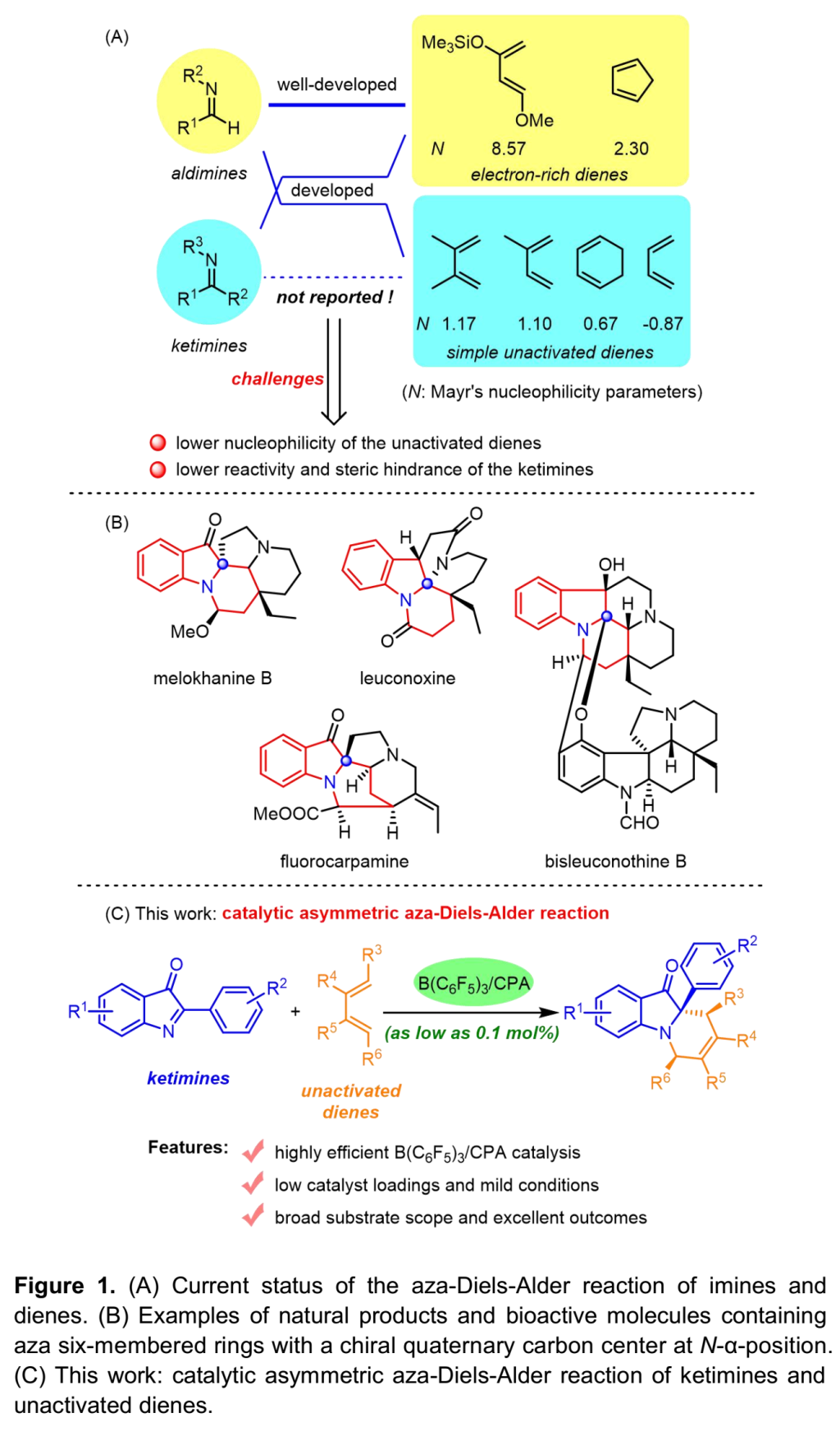
(图片来源:Angew. Chem. Int. Ed.)
首先,作者采用2-芳基-3H-吲哚-3-酮1a与2,3-二甲基-1,3-丁二烯2a作为模板底物,通过对催化剂、溶剂以及温度等条件的筛选,确定最佳的反应条件 (Table 1)为:在氩气存在下,采用2 mol% B(C6F5)3/(R)-B1作为催化剂,50 mg 3 Å MS作为添加剂,MTBE/n-hexane(v/v = 1/3)作为反应溶剂,反应温度为-30 °C,反应时间为6 h,能够以98%的收率与91%的对映选择性获得手性产物3aa。

(图片来源:Angew. Chem. Int. Ed.)
在上述最佳反应条件下,作者首先对酮亚胺的底物范围进行考察 (Table 2)。研究表明,各种带有吸电子、供电子基取代的酮亚胺均能够较好与上述反应条件兼容,并以优秀的收率与良好至优秀的对映选择性获得相应产物。
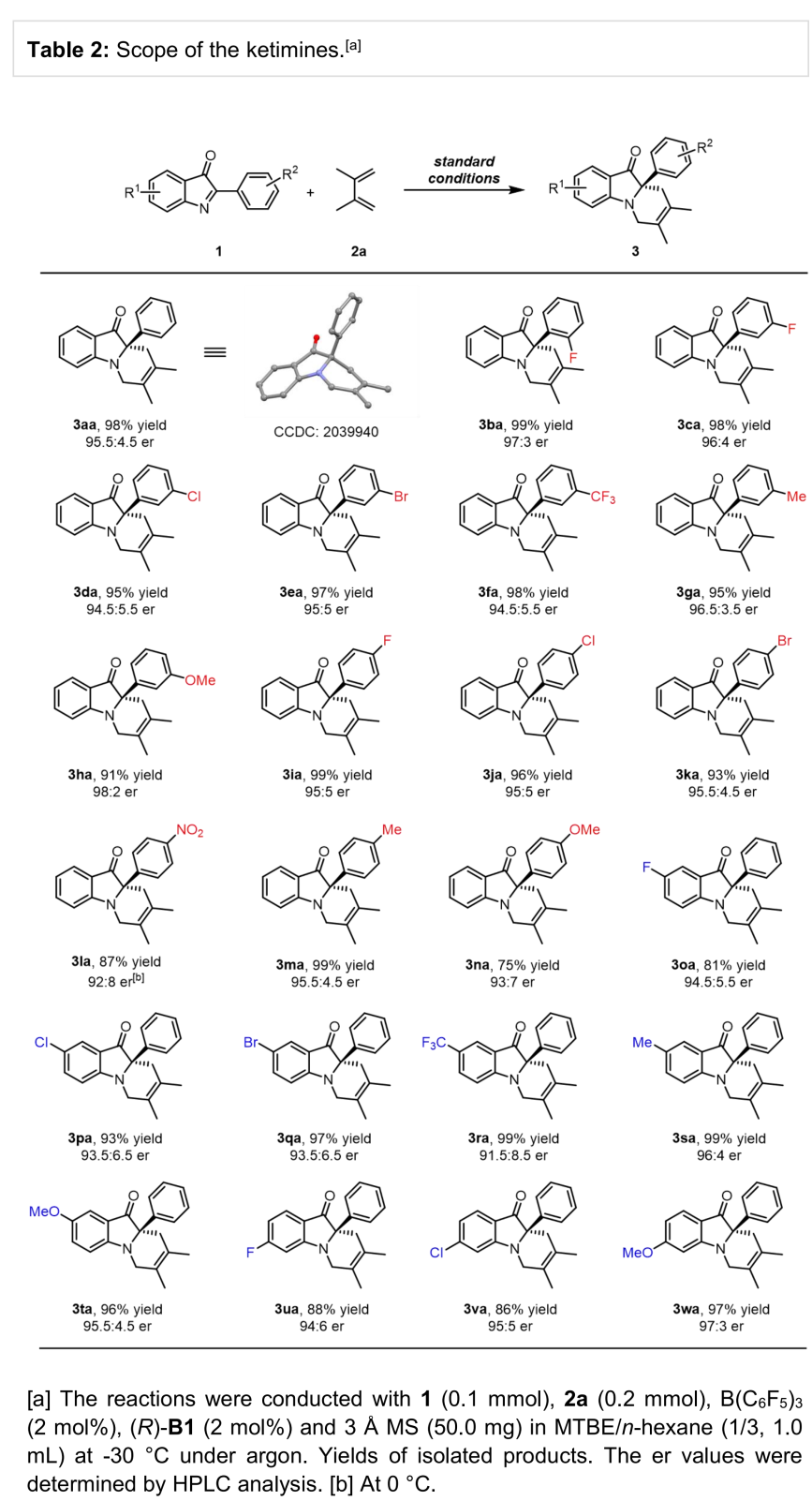
(图片来源:Angew. Chem. Int. Ed.)
接下来,作者进一步考察非活化二烯的底物应用范围 (Table 3)。作者观察到,反应活性较低的1,3-丁二烯 (2b)、简单的2,4-己二烯化合物 (2c)、异戊二烯 (2d)以及β-月桂烯(β-myrcene,2e)等天然二烯与环二烯底物均能够与上述反应条件良好的兼容,并以优秀的收率与良好至优秀的对映选择性获得相应产物。此外,作者同样对官能团化二烯底物的应用进行考察,研究发现,反应同样能够以优秀的收率与非对映选择性获得相应产物,然而,反应过程的对映选择性出现较为显著地降低。
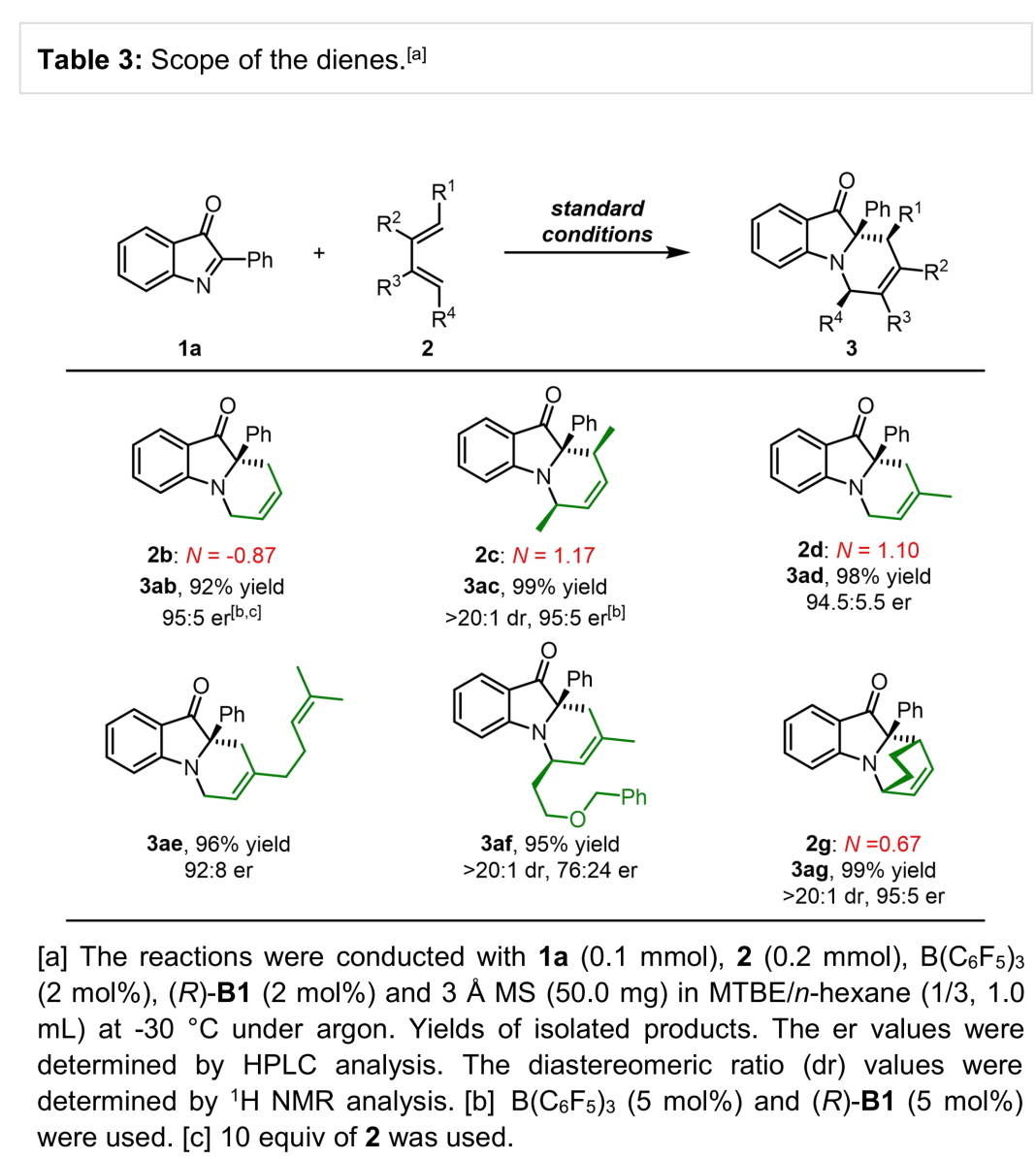
(图片来源:Angew. Chem. Int. Ed.)
为进一步阐明该方法学在有机合成中的应用潜力,作者进行了一系列克级规模反应与衍生化反应 (Scheme 1)。首先,该小组发现,将1a的用量扩大至4.5 mmol,催化剂用量降至0.1 mol%时,反应同样能够以70%的收率与90%的对映选择性获得手性产物3aa (Scheme 1A)。接下来,作者发现,在不同的反应条件下,3aa能够转化为相应的醇类化合物4、末端烯烃5以及含硫化合物6。此外,3ad能够发生硼氢化-氧化反应,转化为醇类化合物7。
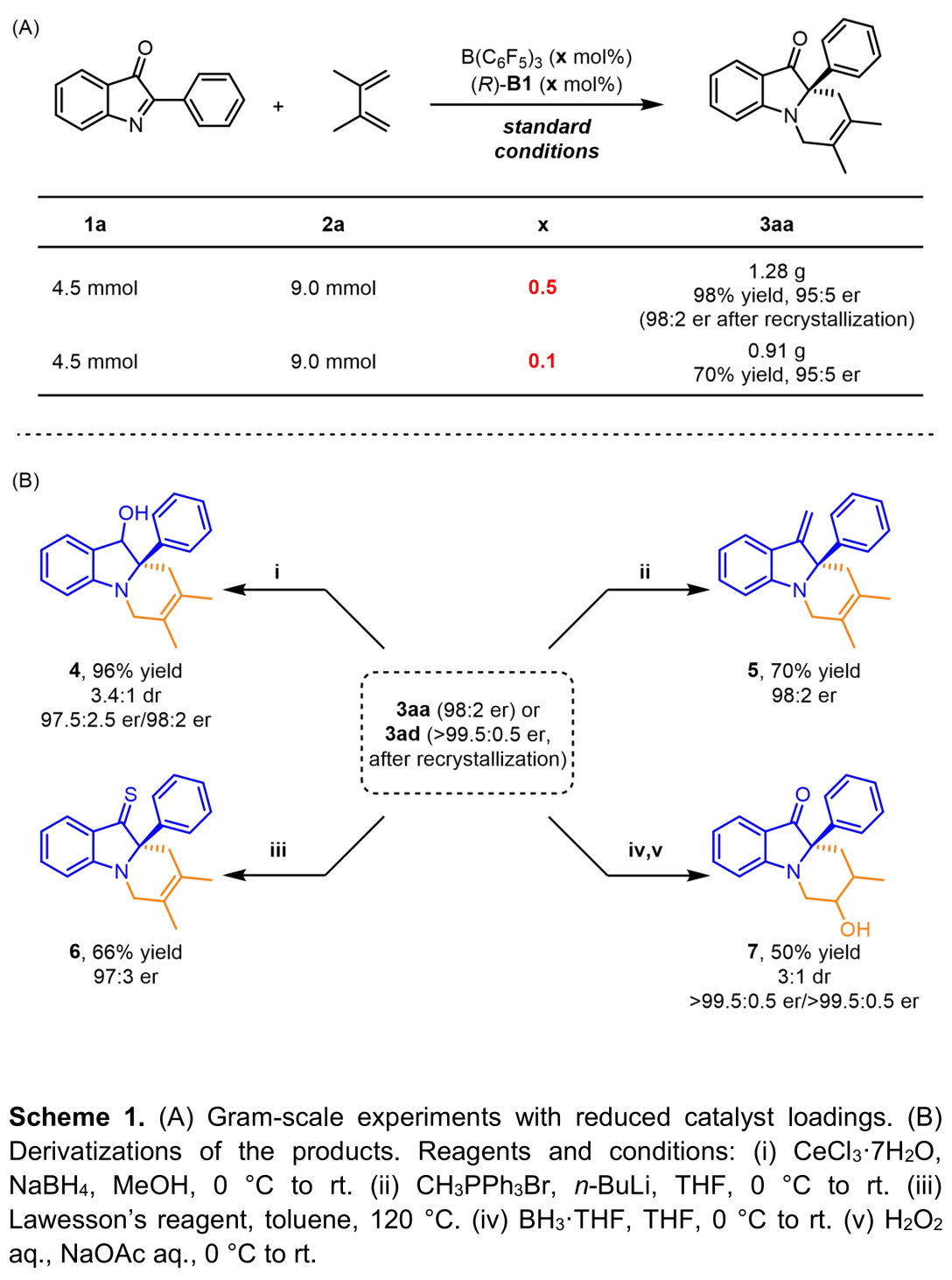
(图片来源:Angew. Chem. Int. Ed.)
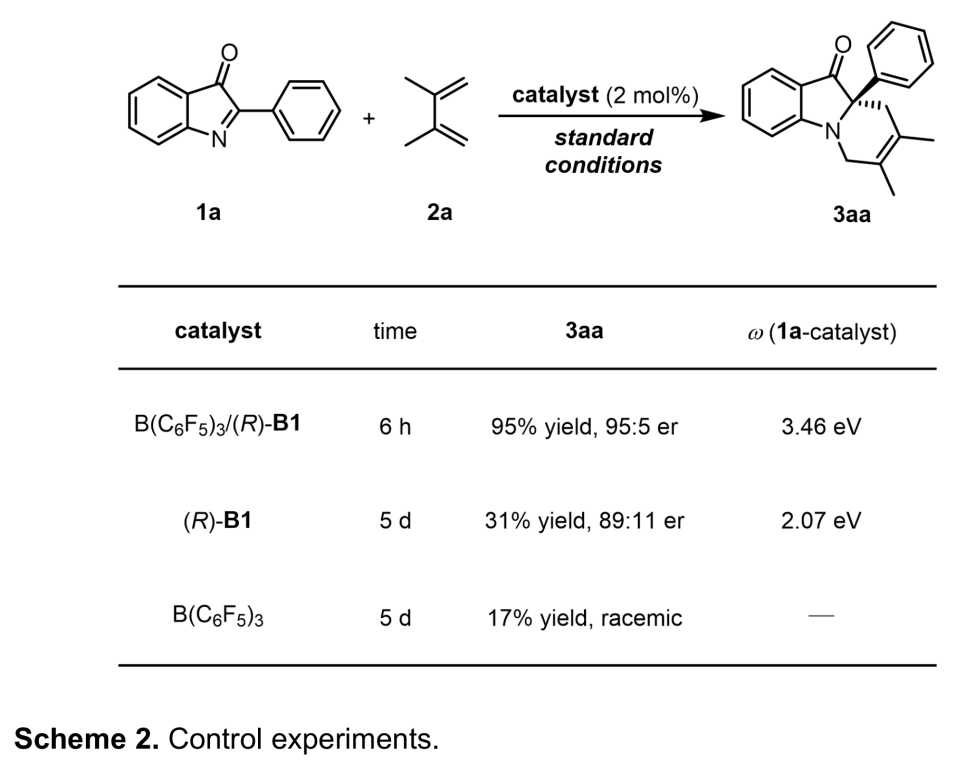
为阐明合理的反应机理,作者进行了一系列相关的控制实验 (Scheme 2)。首先,标准反应条件下,在不加入B(C6F5)3的情况下,作者观察到,反应产物的收率与对映选择性出现显著下降。接下来,作者发现,在未加入手性磷酸的情况下,反应收率急剧下降。由此可以推测B(C6F5)3与CPA的协同效应 (cooperative effect)可能源自于CPA与B(C6F5)3配位之后,相应酸性的增强。而且,计算结果表明,(R)-B1与B(C6F5)3配位之后,其pKa值由5.0 降低至-4.4[5]。综上结果,作者推测,(R)-B1/B(C6F5)3催化体系较强的酸性可能成为提高反应速率的关键因素。同时,对1a亲电参数 (ω)的计算支持上述推测。计算发现,在采用(R)-B1/B(C6F5)3进行活化之后,1a的亲电参数由2.07 eV显著增加至3.46 eV,这表明通过(R)-B1/B(C6F5)3的活化,能够显著提高1a的反应活性。
(图片来源:Angew. Chem. Int. Ed.)
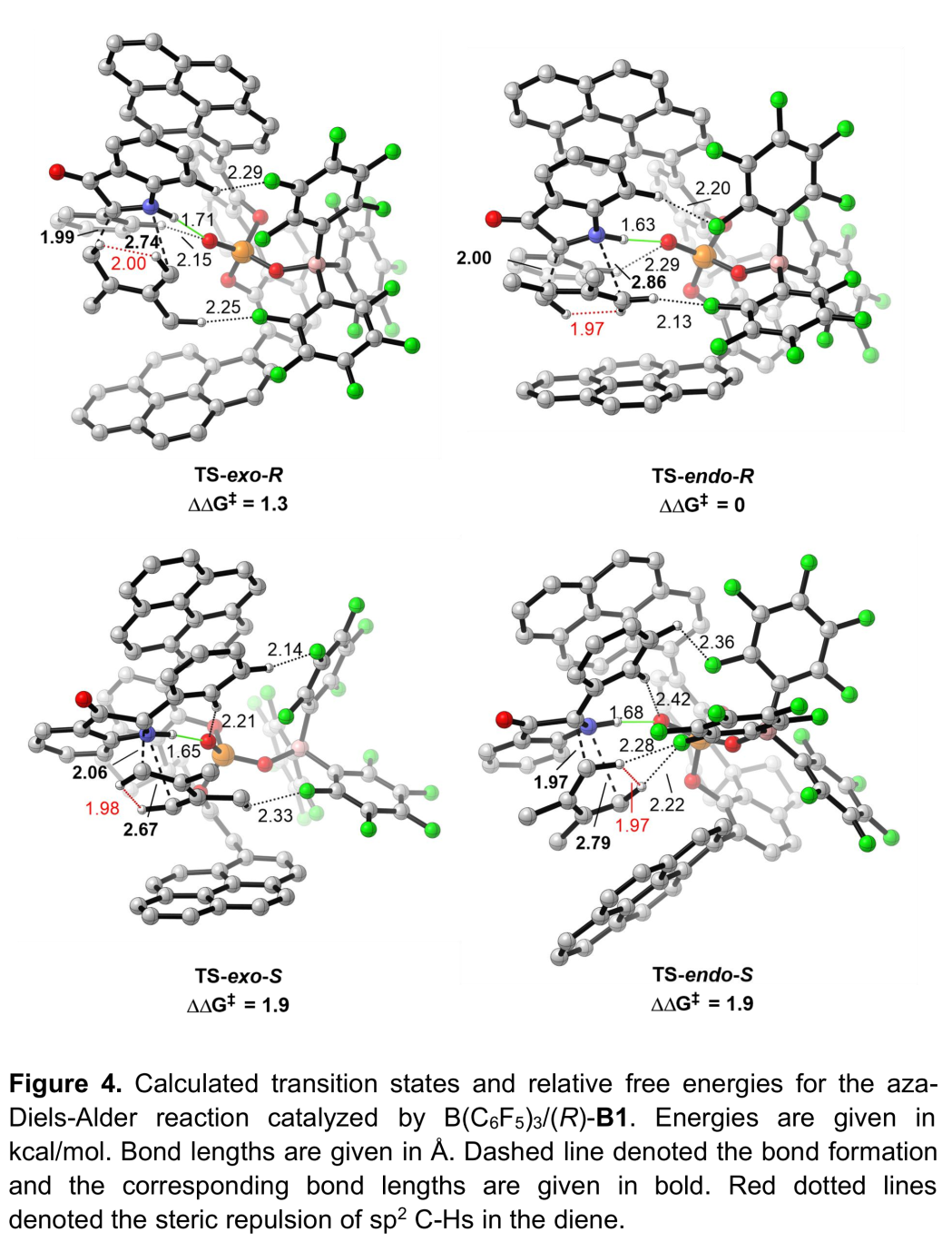
为进一步研究影响反应过程立体选择性的相关因素,作者进行相应的DFT计算(Figure 4)。计算结果表明次级轨道相互作用 (secondary orbitalinteraction, SOI)与非共价相互作用 (non-covalent interaction, NCI)为控制反应过程立体选择性的关键因素。此外,作者进一步推测,通过B(C6F5)3中F原子产生的弱相互作用,例如,C-H•••F氢键作用,同样可能成为控制反应立体选择性的重要因素。
(图片来源:Angew. Chem. Int. Ed.)
小结
南开大学李鑫教授团队发展出通过B(C6F5)3/CPA催化体系参与的酮亚胺与非活化二烯之间的不对称氮杂Diels-Alder反应方法学,并能够以优秀的收率与对映选择性获得一系列含有季碳手性中心的四氢吡啶类化合物。此外,作者通DFT计算研究阐明反应过程中控制立体选择性的关键因素为次级轨道相互作用与非共价相互作用。本文第一作者为南开大学博士研究生赵群。

















No comments yet.