本文作者:杉杉
导读
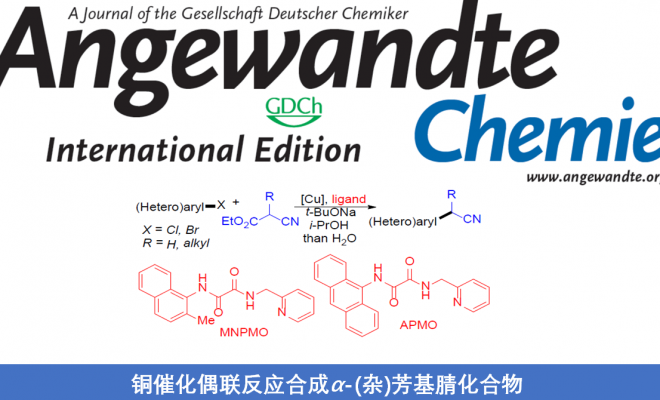
α-(杂)芳基腈作为药物分子设计中常见的结构单元,然而,常规的通过与(杂)芳卤之间的偶联过程,合成α-(杂)芳基腈的方法学中,底物适用范围较为有限。近日,上海有机所的马大为教授课题组在Angew. Chem. Int. Ed.中发表论文,报道了将铜盐与草酸二酰胺(oxalic diamides)配体MNPMO(N-(2-methylnaphthalen-1-yl)-N’-(pyridin-2-ylmethyl)oxalamide)结合,从而在温和的反应条件下,顺利完成各类(杂)芳卤(Cl,Br)与氰乙酸乙酯(ethyl cyanoacetate)之间的偶联反应,进而获得一系列α-(杂)芳基腈衍生物。同时,作者发现,采用CuBr/草酸二酰胺配体APMO(N-( anthracenyl)-N’-(pyridin-2-ylmethyl)oxalamide)体系,能够进一步完成(杂)芳基溴与α-烷基取代的氰乙酸乙酯的偶联反应,获得一系列α-烷基(杂)芳基乙腈衍生物。
Assembly of α-(Hetero)aryl Nitriles via Cu-Catalyzed Coupling Reactionswith (Hetero)aryl Chlorides and Bromides
Y.Chen, L Xu, Y Jiang,D. Ma,
Angew. Chem. Int. Ed. ASAP. DOI:10.1002/anie.202014638.
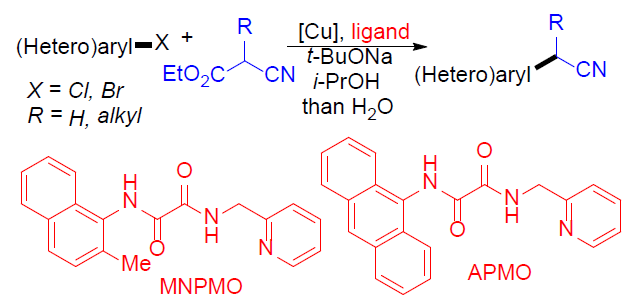
正文
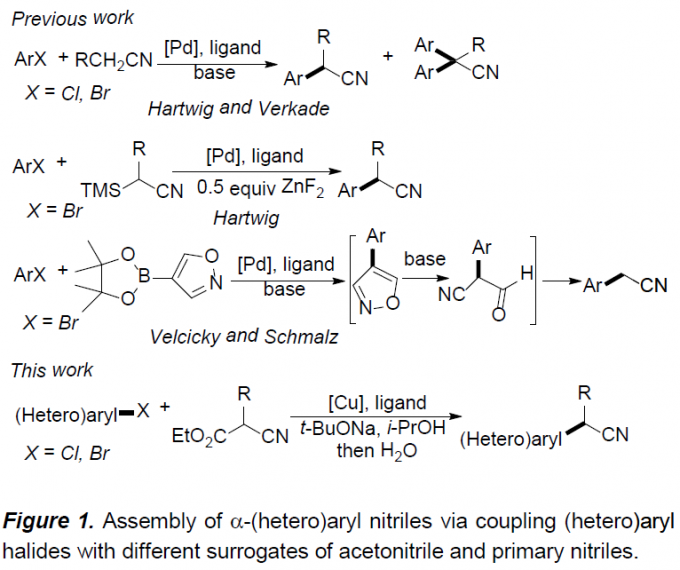
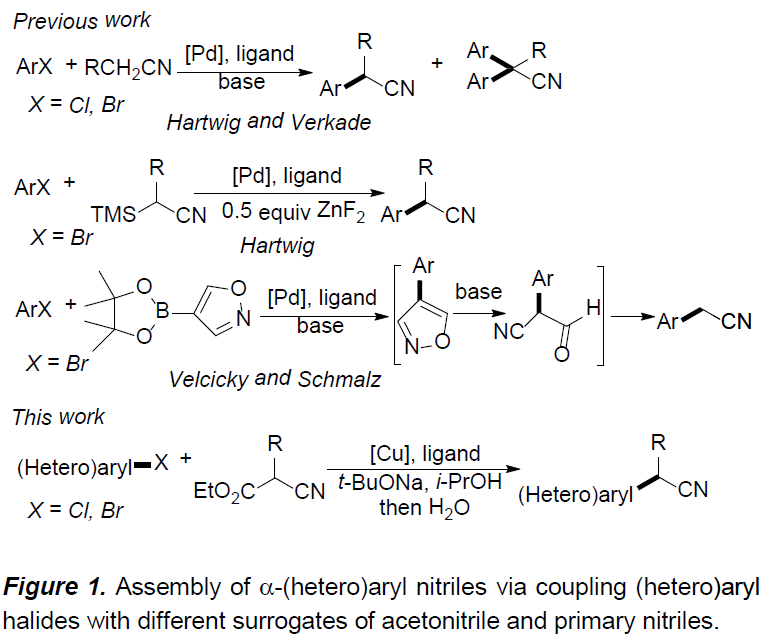
α-(杂)芳基腈作为一系列药物分子中重要的结构单元,以及有机合成中的重要砌块,尤其在制药工业中(主要涉及非甾体抗炎药(NSAID, non-steroidal anti-inflammatory drug)的生产),常用作制备α-芳基酰胺与羧酸的起始原料。在过去的二十年中,通过选择不同的偶联策略,已开发出多种合成α-芳基腈[1-4],α-芳基羧酸[5],α-芳基酯[6]以及酰胺[7]的反应方法学。其中,金属催化的芳基卤与腈之间的偶联反应,由于相应偶联试剂廉价易得,进而在工业大规模合成(large-scale synthesis)中具有良好的应用前景。同时,Hartwig[1a]与Verkade[1b]报道了通过钯盐与位阻膦配体催化下,简单腈的直接芳基化反应方法学(Figure 1)。然而,在选择一级腈参与上述反应时,大多数情况下,会获得相应腈单芳基化与二芳基化产物的混合物。为了解决这一问题,Hartwig与Wu等[1c]选用α-甲硅烷基腈代替腈作为偶联试剂,从而成功实现乙腈与一级腈的选择性单芳基化。而Velcicky等[2]采用异噁唑基团作为氰亚甲基单元的前体,通过domino Suzuki偶联-异噁唑碎片化过程,进而获得一系列α-芳基乙腈化合物。尽管Hartwig的报道中,仅对各类杂芳基溴底物的应用范围进行考察,而未考察其它底物。 然而,鉴于这一策略在药物开发过程中表现出的良好应用价值,因此,更加需要发展一种能够多样化构建α-(杂)芳基腈分子的有效策略。在过去的几十年中,金属催化的芳卤与氰乙酸乙酯间的偶联方法学备受关注[9-11]。然而,上述方法学的应用同样存在一定限制,例如,Cu催化的芳基化仅能够应用于芳基碘底物 [9,10],而Pd催化的芳基化中采用昂贵的金属与配体,同时,反应条件难以应用于多数的杂芳卤以及α-烷基取代的氰乙酸乙酯 [11]。这里,上海有机所的马大为课题组报道了通过将铜盐与草酸二酰胺配体结合,进而使各种(杂)芳基卤(Cl,Br)与氰乙酸乙酯在温和的反应条件下进行偶联,最终获得一系列α-(杂)芳基腈衍生物。
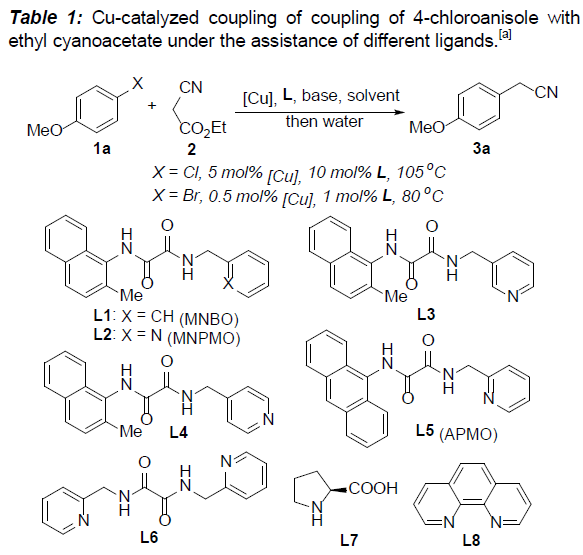
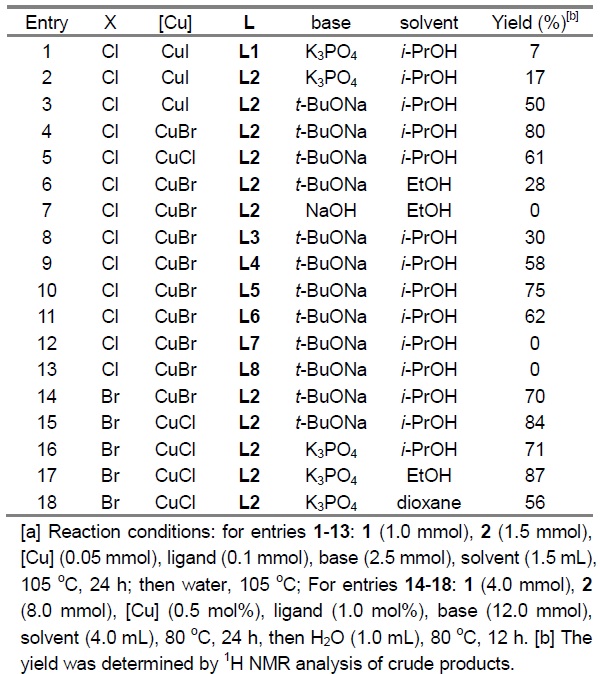
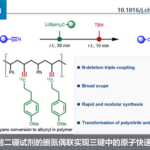
基于前期草酸二酰胺配体促进的铜催化偶联反应的研究[12],作者选择4-卤代苯甲醚(1)与氰基乙酸乙酯(2)作为模型底物,对相关反应条件进行优化筛选(Table 1)。研究发现,在选择 4-氯苯甲醚作为反应底物时,采用5 mol%的CuBr作为催化剂,10 mol%的L2 (MNPMO)作为配体,t-BuONa作为碱,并在异丙醇溶剂中,105℃下进行反应,最终获得80%收率的产物3a。同时,作者进一步表明,在选择4-溴苯甲醚作为反应底物时,采用0.5 mol%的CuCl作为催化剂,1 mol%的L2作为配体,K3PO4作为碱,在乙醇溶剂中,80℃下进行反应,能够获得87%收率的产物3a。
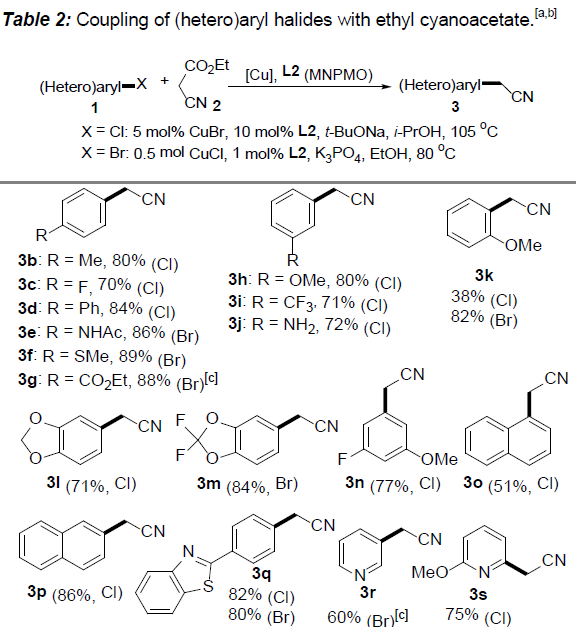
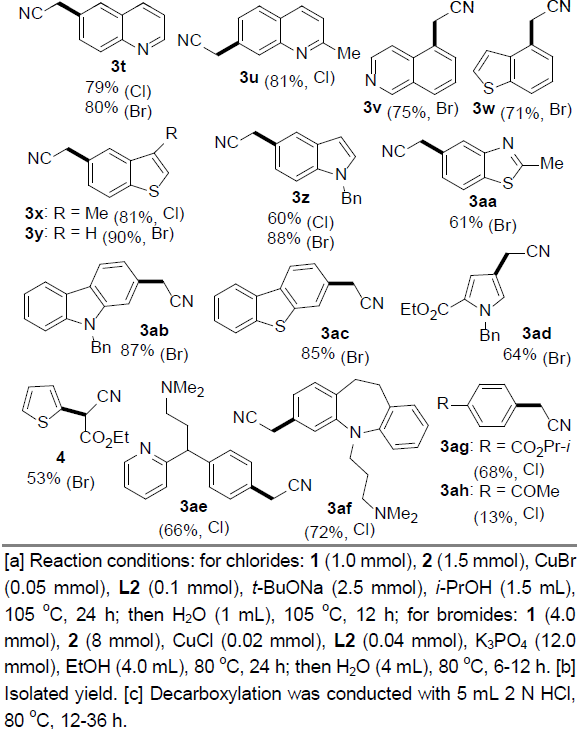
在获得上述最佳反应条件后,作者开始对(杂)芳卤1的应用范围进行考察(Table 2)。研究发现,对位与间位带有供电子或吸电子基团的芳卤(Cl,Br),均能够顺利完成上述反应,并以70-89%收率获得相应的产物3b–3j。值得注意的是,在选择4-溴苯甲酸乙酯作为反应底物时,为保留产物3g中的酯基官能团,需要在酸性条件下进行后续的脱羧过程。之后,作者观察到,2-氯苯甲醚参与的偶联过程,获得产物3k的收率较低,然而,与2-溴苯甲醚偶联过程,却能够获得良好的反应收率。这些事实充分表明立体位阻对芳基氯参与的偶联过程具有显著影响。此外,作者通过对2-氯萘(3p)与1-氯萘(3o)参与上述偶联过程时,产物收率差异的研究,进一步阐明立体位阻对芳基氯底物参与偶联过程的显著影响。其次,作者观察到上述反应条件对于三取代的芳卤与双重杂环取代的芳卤同样能够良好地兼容,并以较高的反应收率获得产物31–3n。随后,作者发现,在上述标准反应条件下,一系列杂芳基卤(Cl,Br)的偶联反应均能够顺利进行,从而,以良好至优良的收率获得相应偶联产物3r–3ad。同时,作者进一步研究表明,上述方反应条件对于已知的含有芳基氯结构单元的药物分子同样适用。并能够顺利可实现这类分子的直接官能团化,例如chlorpheniramine与clomipramine的偶联反应,分别获得产物3ae与3af。另外,作者发现,选用带有酯基取代的芳基氯底物时,能够以68%的收率获得相应产物3ag,而选择乙酰基取代的芳基氯底物时,却以极低的收率获得产物3ah。
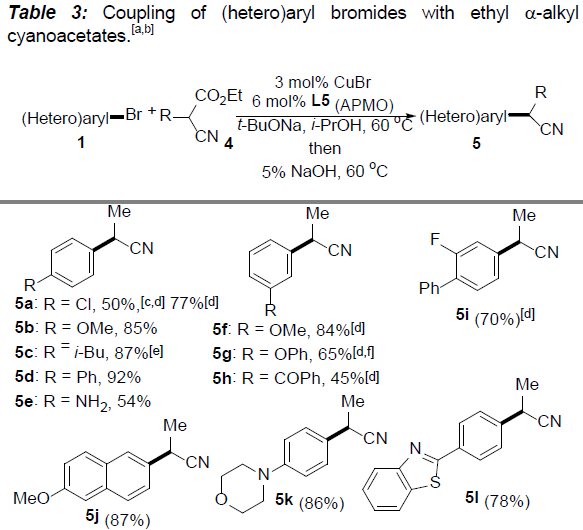
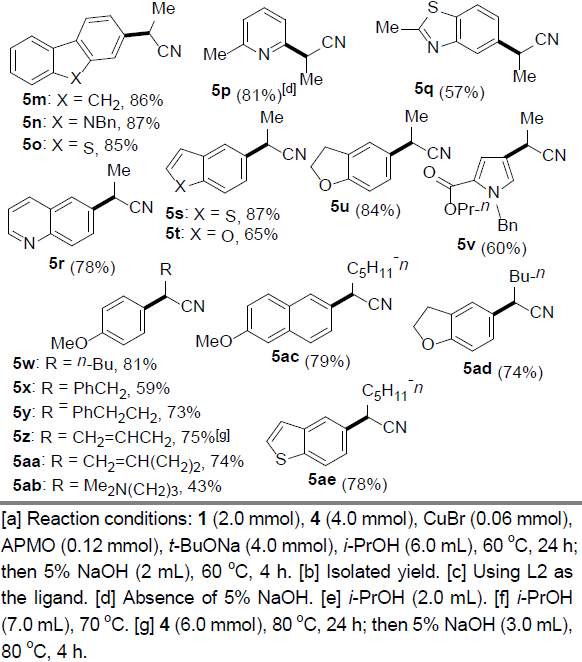
接下来,作者在对α-烷基取代的氰乙酸乙酯的适用范围进行考察时发现,在上述标准条件下,仅能够获得50%的收率的目标产物5a,然而,作者在选择L5 (APMO)作为配体时,却能够将收率提高至77%。通常,在偶联反应完成之后,产物后续的脱羧反应为自发过程。然而,尤其对于带有供电子基团取代的偶联产物,则无法有效地完成脱羧过程。此时,则需要加入5%NaOH,并延长加热时间,进而获得较高收率的目标产物。
同时,作者对(杂)芳基溴与α-烷基取代的氰乙酸乙酯之间偶联反应的底物应用范围进行深入研究(Table 3)。结果表明,一系列官能化的(杂)芳基溴,均能够顺利完成上述偶联反应,并获得相应产物5a–5v。除2-氰基丙酸乙酯之外,其它具有更高立体位阻的类似底物同样能够与上述反应条件良好地兼容,并以43-81%的收率获得相应目标产物5w–5ad。此外,氰乙酸乙酯侧链中烯基(5z,5aa)与氨基官能团(5ab)的存在,无法改变上述偶联过程。
值得注意的是,上述反应的部分目标产物能够成功应用于一系列NSAID药物的合成,例如Ibuprofen(5c起)、Fenoprofen(5g起)、Ketoprofen(5h起)、Flurbiprofen(5i起)、Naproxen(起5j)、Cicloprofen(5m起)、Indoprofen(5e起)、Araprofen(5e起)以及Alminoprofen(5e起)。因此,该方法学的发展为上述的已知药物及其类似物分子的构建提供了一种简便易行的途径。
总结
上海有机所马大为课题组报道了通过将铜盐与草酸二酰胺配体(L2与L5)结合,从而成功实现(杂)芳基卤化物与氰基乙酸乙酯及其α-单烷基化衍生物之间的偶联反应方法学,进而获得一系列α-(烷基)(杂)芳基腈衍生物。同时,该方法学首次选用廉价易得的(杂)芳基氯与(杂)芳基溴作为偶联底物。此外,上述方法学具有良好的官能团兼容性,能够应用于合成各类取代α-(杂)芳基腈衍生物。
参考文献
[1] a) D. A. Culkin, J. F. Hartwig,
J. Am. Chem. Soc.
2002,
124, 9330. b) J. You,J. G. Verkade,
Angew. Chem. Int. Ed. 2003,
42, 5051.
Angew.Chem.2003,
115, 5205. c) L. Wu, J. F. Hartwig,
J. Am. Chem. Soc.
2005,
127, 15824.
[2] a) J. Velcicky, A. Soicke, R. Dteiner, H.-G. Schmalz,
J. Am. Chem.Soc.
2011,
133, 6948. For a similar study, see: b) P. J. Lindsay-Scott, A. Clarke, J. Richardson,
Org. Lett.
2015,
17, 476.
[3] For selected references on Pd-catalyzed coupling of aryl halides with α,α-disubstituted nitriles, see: a) S. H. Kim, W. Jang, M. Kim, J. G.Verkade, Y. Kim,
Eur. J. Org. Chem.
2014, 6025. b) Z. Jiao, K.W. Chee, J. Zhou,
J. Am. Chem. Soc.
2016,
138, 16240. c) B. A.Wright, M. J. Ardolino,
J. Org. Chem.
2019,
84, 4670.
[4] a) R. Shang, D. Ji, L. Chu, Y. Fu, L. Liu,
Angew. Chem. Int. Ed. 2011,
50, 4470.
Angew. Chem.2011,
123, 4562. b) G. Wu, Y.Deng, C. Wu, Y. Zhang, J. Wang,
Angew. Chem. Int. Ed. 2014,
53, 10510.
Angew. Chem. 2014,
126, 10678.
[5] a) F. Mandrelli, A. Blond, T. James, H. Kim, B. List,
Angew. Chem.Int. Ed. 2019,
58, 11479.
Angew. Chem. 2019,
131, 11603. b) Z.-T. He, J. F. Hartwig,
J. Am. Chem. Soc.
2019,
141, 11749.
[6] a) H. Guan, Q. Zhang, P. J. Walsh, J. Mao,
Angew. Chem. Int. Ed. 2020,
59, 5172.
Angew. Chem. 2020,
132, 5210. b) N. Holmberg-Douglas, N. P. R. Onuska, D. A. Nicewicz,
Angew. Chem. Int. Ed. 2020,
42, 7425.
Angew. Chem.2020,
132, 7495. c)T. Q. Chen, D. W. C. Mac Millan,
Angew. Chem. Int. Ed. 2019,
58, 14584.
Angew. Chem. 2019,
131, 14726.
[7] B. Li, T. Li, M. A. Aliyu, Z. H. Li, W. Tang,
Angew. Chem. Int. Ed. 2019,
58, 11355.
Angew. Chem. 2019,
131, 11477.
[8] a) J. J. Baldwin, S. Cacatian, D. Claremon, L. W. Dillard, P. T. Flaherty, B. Ghavimi-Alagha, D. Ghirlanda, A. V. Ishchenko, L. S. Kallander, B. Lawhorn, Q. Lu, G. Mcgeehan, L. R. Terrell, J. Zhang, C. A. Leach, P. Stoy, J. R. Patterson, R. D. Simpson, S. B. Singh, C.Tice, Z. Xu, C. C. K. Yuan, W. Zhao, J. Yuan, WO2008124575(2008). b) J. Zhuo, M. Xu, C.He, C. Zhang, D. Qian, B. Metcalf, W.Yao, US Patent 7767675 (2010). c) H. R. Chobanian, Y. Guo, P. Liu,T. J. Lanza, M. Chioda, L. Chang, T. M. Kelly, Y. Kan, O. Palyha, X.-M. Guan, D. J. Marsh, J. M. Metzger, K. Raustad, S.-P. Wang, A.M. Strack, J. N. Gorski, R. Miller, J. Pang, K. Lyons, J. Dragovic, J. G.Ning, W. A. Schafer, C. J. Welch, X. Gong, Y.-D. Gao, V. Hornak, M. L. Reitman, R. P. Nargund, L. S. Lin,
Bioorg. Med. Chem.
2012,
20, 2845. d) B. O. Buckman, J. B. Nicholas, K. Emayan, S. D. Seiwert, S. Yuan, US20140213538 (2014). e) A. Edmunds, M.Muehlebach, P. J. M. Jung, A. Jeanguenat, A. Buchholz, US Patent 10435401 (2019).
[9] a) A. Osuka, T. Kobayashi, H. Suzuki,
Synthesis 1983, 67. b) H.Suzuki, T. Kobayashi, Y. Yoshida, A. Osuka,
Chem. Lett.
1983, 193. c) K. Okuro, M. Furuune, M. Miura, M. Nomura,
J. Org.Chem.
1993,
58, 7606. d) H. J. Cristau, P. P. Cellier, J. F. Spindler, M, Taillefer,
Chem. Eur. J.
2004,
10, 5607. e) Y. Jiang, N. Wu, H. Wu, M. He,
Synlett 2005,
18, 2731. f) S. Xie, P. Qin, M. Li, X. Zhang, Y. Jiang, D. Ma,
Tetrahedron lett.
2013,
54, 3889.
[10] For a recent review, see: S. Bhunia, G. G. Pawar, S. V. Kumar, Y.Jiang, D. Ma,
Angew. Chem. Int. Ed. 2017,
56, 16136.
Angew.Chem. 2017,
129, 16325.
[11] a) M. Uno, K. Seto, W. Ueda, M. Masuda, S. Takahashi,
Synthesis,
1985, 506. b) S. R. Stauffer, N. A. Beare, J. P. Stambuli, J. F. Hartwig,
J. Am. Chem. Soc.
2001,
123, 4641. c) N. A. Beare, J.F. Hartwig,
J. Org. Chem.
2002,
67, 541. d) J. You, J. G. Verkade,
J. Org. Chem.
2003,
68, 8003. e) C. Gao, X. Tao, Y. Qian, J. Huang,
Synlett 2003,
11, 1716. f) P. Y. Yeung, K. H. Chung, F. Y. Kwong,
Org. Lett.
2011,
13, 2912. g) N. Todorovic, E. Awuah, S. Albu, C. Ozimok, A. Capretta,
Org. Lett.
2011,
13, 6180. h) S. H. Kim, W. Jang, M. Kim, J. G. Verkade, Y. Kim,
Eur. J. Org. Chem.
2014, 6025. i) J. G. Semmes, S. L. Bevans, C. H. Mullins, K. H. Shaughnessy,
Tetrahedron Lett.
2015,
56, 3447.
[12] a) W. Zhou, M. Fan, J. Yin, Y. Jiang, D. Ma,
J. Am. Chem. Soc. 2015,
137, 11942. b) M. Fan, W. Zhou, Y. Jiang, D. Ma,
Org. Lett.2015,
17, 5934. c) S. Xia, L. Gan, K. Wang, Z. Li, D. Ma,
J. Am.Chem. Soc. 2016,
138, 13493. d) M. Fan, W. Zhou, Y. Jiang, D. Ma,
Angew. Chem. Int. Ed. 2016,
55, 6211.
Angew.Chem. 2016,
128, 6319. e) J. Gao, S. Bhunia, K. Wang, L. Gan, S. Xia, D. Ma,
Org. Lett. 2017,
19, 2809. f) Z. Chen, Y. Jiang, L. Zhang, Y. Guo, D. Ma,
J. Am. Chem. Soc. 2019,
141, 3541.

















No comments yet.