
本文投稿作者 大白菜
Bryostatin 1是正在临床试验中的消灭艾滋病毒/艾滋病的第一类潜伏期逆转剂。它被用来治疗阿尔兹海默症,也可以作为抗癌症的免疫治疗剂(1-4)。这些疾病是导致死亡的主要原因,影响了成千上万的病人和护理人员。Bryostatin1的假定目标蛋白激酶C(PKC),PKC在临床前研究中表明其与各种未得到满足的神经和心血管适应症密切相关(5)。
尽管目前有潜在的临床应用价值,美国国立癌症研究所(NCI)有限的Bryostatin 1(原为18克)在提供肿瘤学和阿尔茨海默氏病的40多个临床试验之后,它几乎用尽了。Bryostatin 1的长期稀缺严重限制了其研究和临床研究,也妨碍了获得潜在优秀的衍生物和类似物。
自从1968年首次分离以来,人们一直在寻找一些解决Bryostatin供应问题的方法。NCI原来的“手工收集”14吨海洋生物 Bugula neritina只提供了18克Bryostatin1(0.00014%收率)。虽然有报道合成生物学的方法,但由于与Bryostatin生产相关的共生细菌的培养是困难的,所以仍处于初期阶段。多年来,Bryostati 家族的各种成员的化学合成已经改善,从多达90步到少至36步。 唯一报道的Bryostatin 1的合成需要57步(6)。
近期Paul A. Wender课题组(主页)报道了一种以步骤经济,克级规模的合成方法:
Scalable synthesis of bryostatin 1 and analogs, adjuvant leads against latent HIV
Paul A. Wender, Clayton T. Hardman, Stephen Ho, Matthew S. Jeffreys, Jana K. Maclaren, Ryan V. Quiroz, Steven M. Ryckbosch, Akira J. Shimizu, Jack L. Sloane, Matthew C. Steven
Science, 2017, 358, 218–223 , DOI: 10.1126/science.aan7969
该方案解决Bryostatin 1供应问题,也同时满足临床和研究需求,并可作为探索新的和潜在优越的类似物的实用平台。作者一共汇聚合成了29步,最长线性序列(LLS)19步,总收率4.8%(每步平均产率> 80%),并共生产了2克Bryostatin 1。除了出于安全原因的最后一步之外的所有步骤,都是以克到数克的规模进行的。因此,这种合成可以容易地提供大量材料以进一步推进临床评估。因为根据目前使用的临床剂量,单克Bryostatin 1可以治疗约1000个癌症患者或约2000个阿尔茨海默病患者。
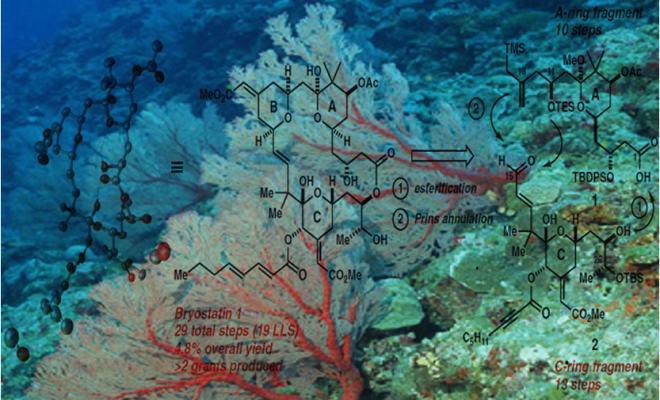
Bryostatin 1构成的合成挑战部分其中的大环内酯结构,它包含三个嵌入氢化吡喃环,十一个立构中心,以及多个烯烃,醇,醚,半缩酮和酯官能团的强大阵列。作者应对这一挑战的方法(Fig. 1)受到了他们早期Byostatin 9和类似物合成的启发,设计去合成类似却不复杂的目标子片段A环和C环。这些前体通过Yamaguchi酯化和Prins大环化连接以产生B环,这种聚合策略的另外一个优点是可以修改亚单位或末级中间体以获得衍生物和类似物。
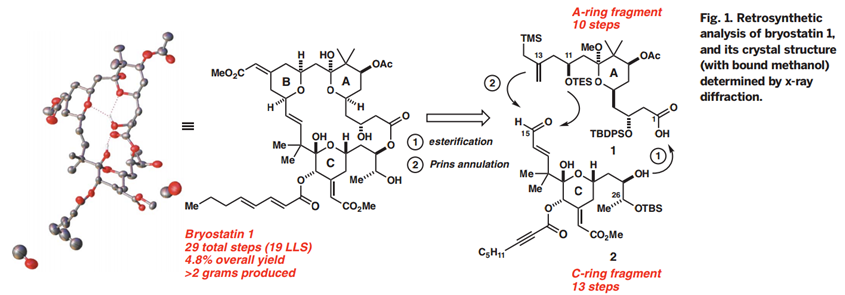
(Fig.1.Retrosynthetic analysis of Bryostatin1)
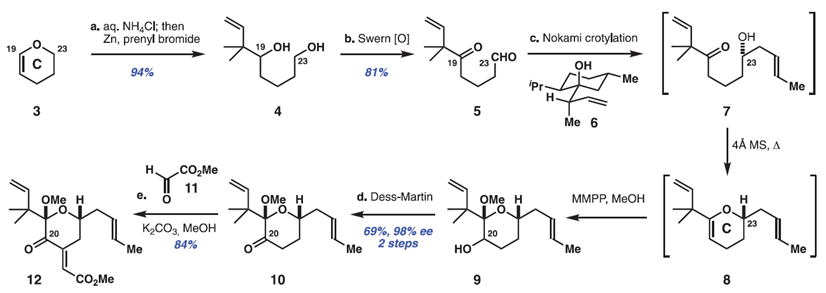
Bryostatin 1中C环片段的合成,其包含了大环的C15至C26部分和影响其PKC亲和力的重要结构单元,从廉价二氢吡喃3($ 3 / mol)的水解和原位异戊烯化开始,形成已知的二醇4(> 20g规模)。这个及随后的所有反应都是由多个研究人员在多个尺度上进行的,以确保可重复性和信息传递。此外,尽管所有可分离的产物都经过纯化和表征,但是设计了许多步骤而不进行产物纯化,以减少时间,成本和产生废物的色谱分析。因此,将粗制二醇4双重氧化,得到1,5-酮醛5(> 25g级,81%收率),开始C环形成阶段。虽然以前尚未探索二羰基化合物,作者发现涉及酮醛5和巴豆基转移试剂6的Nokami的巴豆酰化可以高化学和对映选择性完成醇7中C23立体化学 [> 98%(ee)]。该过程可以一锅法与原位环脱水和烯醇醚环氧化一起进行,以2:1的C20差向异构体的混合物形式得到吡喃9,吡喃9是不稳定的,因此被直接氧化以提供可分离的C20酮10(从化合物5开始的收率是69%),化合物10是在此路线中需要色谱纯化的第一个中间体。
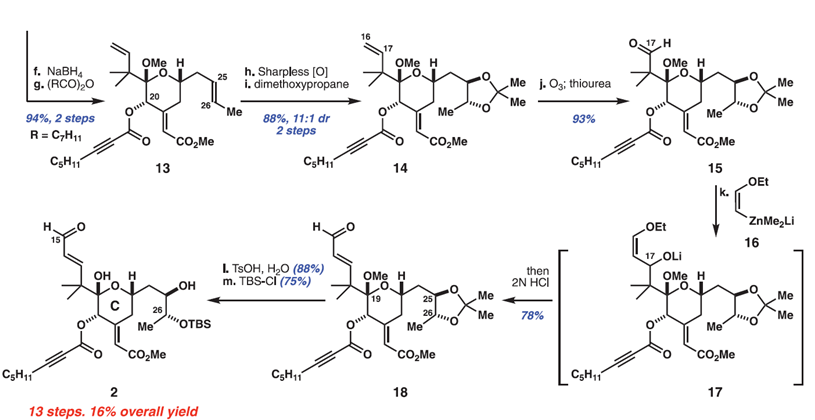

通过一步羟醛缩合反应引入Bryostatin 1中C环烯酸酯,以84%的收率得到专一的E-异构体。然后选择性地还原C20的酮并酯化得到辛炔酸酯13。在其他烯烃和炔烃的存在下,作者首先对化合物13中C25/C26烯烃进行选择性氧化,以Sharpless金鸡纳碱体系,该反应以显著地立体,区域和化学选择性地氧化其中一个π体系。将粗制二醇直接保护,得到丙酮化合物14,两步收率为88%、dr=11:1。随后,化学计量的臭氧分解反应使C16/C17烯烃选择性地断裂,以93%的收率得到醛15。醛15的同系化是一个长期的化学选择性挑战,因为它的空间位阻(包括C25 / C26保护基产生的意外远程效应)以及涉及不饱和酯部分的竞争性去质子化和Michael加成的可能性。作者尝试了许多非碱性的亲核试剂,在原位水解之后,只有乙烯基锌酸酯16干净地使C17醛得到醛18(产率78%)。随后,脱去C25 / C26缩丙酮和C19缩酮保护基团,接着TBS保护C26醇得到醛2。总体而言,这条到Bryostatin 1的C环片段2的路线以13个步骤(LLS)进行,总产率高达16%。
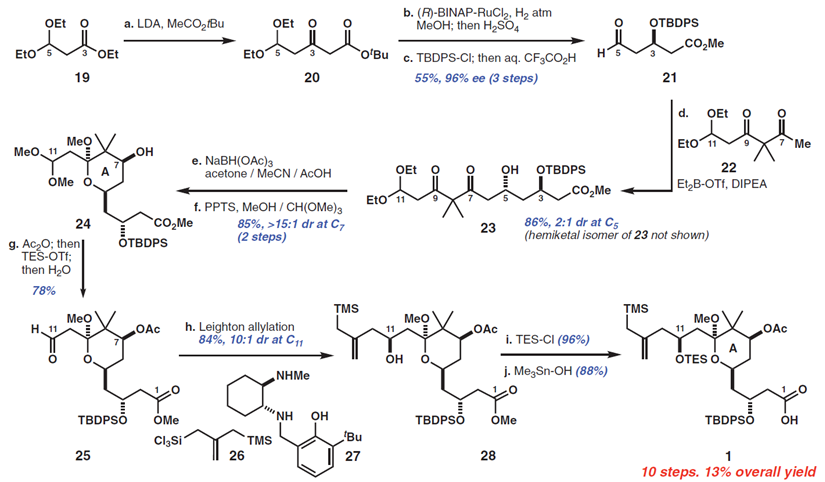

Bryostatin 1的A环片段(Figure 3,acid 1)的合成始于乙酸叔丁酯和丙酸酯19的缩合,罕见的两个烯醇化偶联部分Claisen反应的例子,作者发现,空间位阻大的取代基对亲电试剂(即3,3—二乙氧基基团)与亲核试剂(叔丁基组)来说是必要的,它们可以抑制烯醇交换生成不需要的产品。所得的b-酮酯20用Noyori’s还原催化剂(7)(96%ee),直接转化到醛21。从19到21的这三步通常在30g批次上进行,在一周内进行一次色谱纯化。醛21和β-二酮22进行底物控制的硼羟醛缩合反应得到羟基酮23(收率89%)。酮23的C7位通过改进的Evans-Saksena反应条件(8)进行化学和非对映选择性还原。作者发现应用Me4N-或NaBH(OAc)3在AcOH / MeCN中会导致低的非对映选择性,然而,在引入丙酮作为共溶剂之后,这可能抑制分子间还原,使得dr提高到 >15:1。C9缩酮化(使用原甲酸三甲酯作为脱水剂),然后在C7进行“一锅法”酰化,接着在C11进行化学选择性缩醛水解,然后得到醛25。接着使用Leighton的手性二胺控制剂使醛25和硅26发生非对映选择性烯丙基化反应得到醇28(84%收率,10:1dr)。然后通过C11醇的甲硅烷基化和C1的酯水解完成A环片段1的合成,总收率13%(10步)。
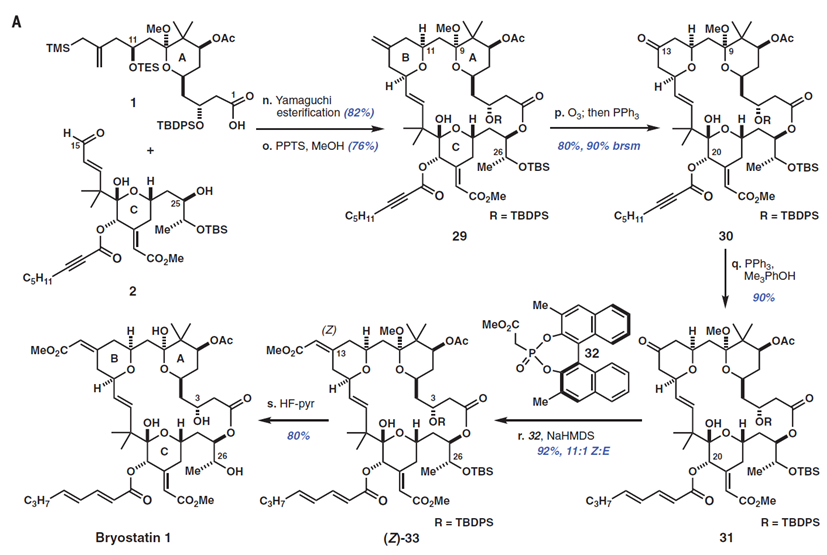
1中A环片段和2中C环片段首先发生Yamaguchi 酯化,然后在甲醇中的4-甲基苯磺酸吡啶(PPTS)催化下,形成B环并关闭大环。所得到的大环内酯包含有4个不同的π体系,这是作者选择性氧化的第三个考验。令人满意的是,29发生化学计量臭氧分解反应并化学选择性产生酮30[80%收率,90%基于回收原料(brsm)],作者发现通过在甲醇存在下进行该反应(与之对比,二氯甲烷),收率从〜50%增加到80%,可能是通过减轻由C9缩酮离子化产生的负面影响。
使用Rychnovsky报道的改进方法,将30的C20炔酸酯用三苯基膦和2,4,6-三甲基苯酚异构化,得到二烯酸酯31,产率90%。作者随后使用Fuji,s膦酸盐32,通过Horner-Wadsworth-Emmons反应来合成B环烯酸酯,作者发现,这种3,3-二甲基-BINOL膦酸酯给出比相应的未被取代的试剂更高的Z:E比率(11:1 vs. 3:1 Z:E)。最后,将33的硅醚和C9缩酮用HF-Py缓冲液裂解,然后原位水解,以80%产率得到Bryostatin 1(从醛2开始总收率30%)。经过高效液相色谱纯化和重结晶(CH2 Cl 2 / MeOH)后,该合成产物纯度> 99.5%。在所有分析数据方面,包括核磁共振和红外光谱,旋光和高分辨率质谱,合成Bryostatin 1与由NCI提供的天然Bryostatin 1的真实样品的数据相同。
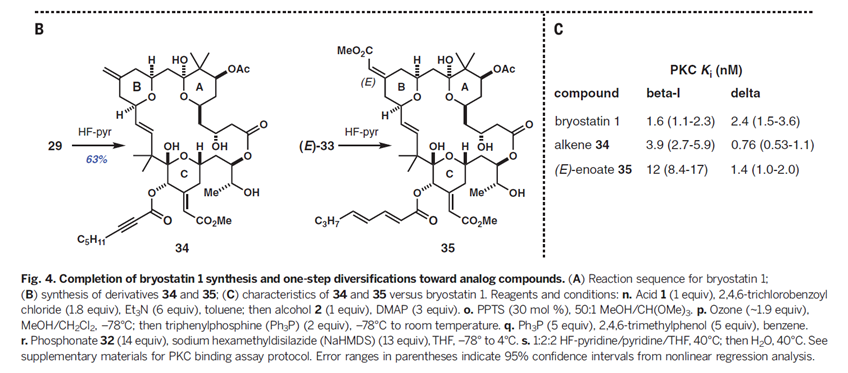
总而言之,该研究打通了一条合成Bryostatin 1及其后期中间体衍生的类似物的克级规模的实用合成路线。例如,具有Bryostatin环结构变化的衍生物34和35(Fig. 4B),可以分别从后期中间体一步反应得到。B环具有潜在的重要性,因为作者的团队最近在长期的(400-500ms)的分子动力学模拟研究中发现,Bryostatin的A和B环施加的氢键网络可以稳定其蛋白质靶标PKC的其他瞬时状态,从而为Bryostatin的独特生物活性提供合理的解释。类似物34和35对PKC-b1和PKC-d(分别代表性的常规和新的PKC同种型)显示出低纳摩尔浓度的亲和力(Fig. 4C)。它们在与细胞的比较研究,动物的疾病模型以及从HIV阳性患者取得的体外样品中也被证明是更有效和具有更好的耐受。我们期望可以看到更多天然产物化学家可以高效高产的合成具有活性的天然产物分子,这也为药物研发开辟的新道路。
参考文献
- 1. T. J. Nelson et al., Alzheimers Dis. 58, 521–535 (2017).
- 2. S. I. Alfonso et al., Signal. 9, ra47 (2016).
- 3. C. Hammond et al., Immunother. 28, 28–39 (2005).
- 4. S. P. Shaha et al., Exp. Immunol. 158, 186–198 (2009).
- D. Mochly-Rosen, K. Das, K. V. Grimes, Nat. Rev. Drug Discov.11, 937–957 (2012).
- G. E. Keck, Y. B. Poudel, T. J. Cummins, A. Rudra, J. A. Covel,J. Am. Chem. Soc. 133, 744–747 (2011).
- J. Nokami et al., J. Am. Chem. Soc. 123, 9168–9169 (2001).
- D. A. Evans, K. T. Chapman, E. M. Carreira, J. Am. Chem. Soc.110, 3560–3578 (1988).
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!

















No comments yet.