
俞寿云教授供稿
导读
近日,南京大学的俞寿云课题组(实验部分)和曲阜师范大学的袁相爱课题组(理论计算部分)合作在J. Am.Chem. Soc.中发表论文,报道首例基于烯烃E → Z异构化的动力学拆分反应。机理验证实验和DFT计算表明,底物−催化剂复合物的两个对映体总体敏化速率的不同,是该异构化过程具有优异动力学拆分效率的决定性因素。该方法进一步拓展了动力学拆分的基态反应模式,为激发态对映选择性催化过程提供一个新的思路。
Photoexcited Chiral Copper Complex-Mediated Alkene E → Z Isomerization Enables Kinetic Resolution. H. Zhang, C. Huang, X. Yuan, S. Yu, J. Am. Chem. Soc. 2022, 144, ASAP. doi: 10.1021/jacs.2c04040.
正文
由于特定烯烃分子的不同异构体(E/Z异构体、对映异构体)对手性受体识别的不同,往往在宏观上表现出不同的物理、化学以及生物学特性。基于此,如何超越生物合成模式的限制,实现烯烃化合物构型的有效控制一直以来都是有机立体化学的重点研究内容。目前,Gilmour[1]、Weaver[2]和俞寿云[3]等课题组利用可见光介导的选择性能量转移策略有效地克服了微观可逆性和基态热力学限制所带来的不利影响,已经能够较好地解决简单烯烃E → Z的有效配置。但是对于三维空间的对映选择性异构化过程来说,则仍然具有十足的挑战。在过去的几十年里,不对称合成已逐步成为构建多样化对映体富集分子的强有力工具。而外消旋混合物的动力学拆分过程,同样也是一种获得手性化合物的成熟策略(Figure 1a)。
近年来,作为有机光化学合成领域中与光氧化还原催化形成互补反应模式的能量转移(energy transfer, EnT)策略,受到了化学家们的广泛关注,并取得了一系列显著的研究成果[4]。然而,三重态能量转移在对映选择性激发态反应中的应用往往会因为手性催化剂和底物之前微弱且不明确的相互作用而受到限制。基于此,开发具有非共价结合位点的三重态手性光敏剂或者双催化策略能够有效克服以上问题(Figure 1b)。受到手性过渡金属配合物(铜[5]、铑[6]、铱[7]等)能够作为激发态不对称催化的光敏剂和手性控制剂的启发,以及最近关于铜配合物能够有效促进烯烃化合物E → Z异构化的报道,南京大学俞寿云课题组(实验部分)近期与曲阜师范大学袁相爱团队(理论计算部分)合作,发展了一种基于光激发手性铜配合物介导的对映选择性E → Z异构化策略,成功实现了一系列外消旋2-苯乙烯基吡咯烷衍生物的动力学拆分(Figure 1c)。
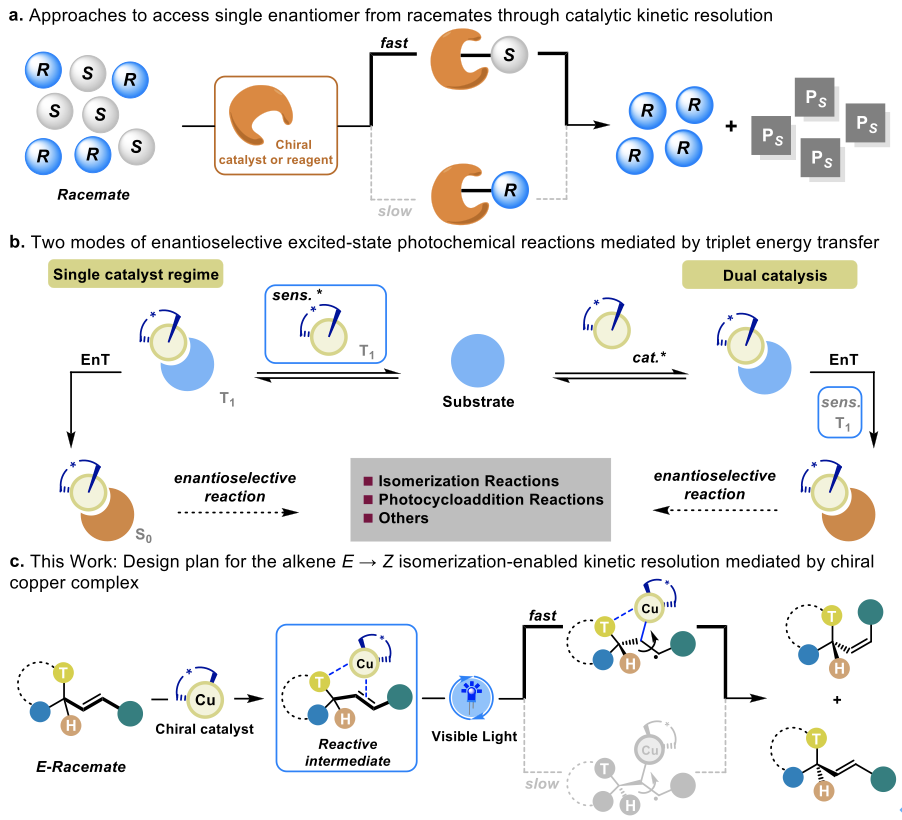
Figure 1. Expanding the potential of kinetic resolution beyond their established ground-state reactivity.
作者首先选定外消旋N-Boc保护的2-苯乙烯基吡咯烷 (±)-E-1a作为模板底物,对该对映选择性异构化过程进行了详细探究。经过一系列反应参数的筛选和优化,他们确定了最佳的反应条件:在二氯甲烷溶剂中,使用Cu(CH3CN)4BF4(5.0 mol%)作为铜催化剂,Box配体L1(7.5 mol%)作为手性配体,通过40 W蓝色LED灯(λmax = 427 nm)的照射,能够顺利地以95% e.e.的对映选择性获得异构化产物 (R,Z)-1a,同时未反应原料 (S,E)-1a也能够以96% e.e.的对映选择性进行回收,该动力学拆分的选择性系数可以高达154。他们也通过一系列控制实验来证明铜催化剂、手性Box配体和光照都是这一对映选择性异构化过程必不可少的,而配体则是决定立体化学结果的唯一因素。
Table 1. Optimization of the kinetic resolution reaction conditionsa
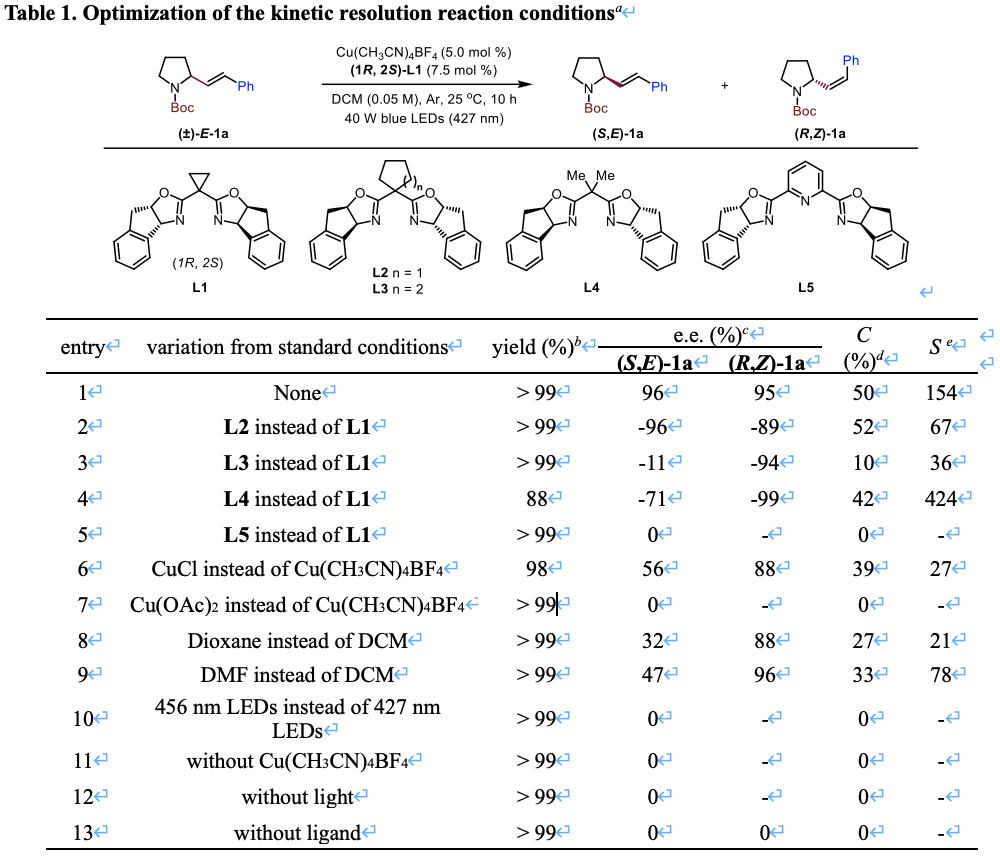
aReactions were carried out with (±)-E-1a (0.05 mmol, E/Z > 20:1), Cu(CH3CN)4BF4 (5.0 mol %) and (1R, 2S)-L1 (7.5 mol %) in DCM (1.0 mL) at 25 oC under 40 W blue LEDs (427 nm) for 10 h. bThe yield was determined by GC in the presence of dodecane as an internal standard. cEnantiomeric excess (e.e.) values were determined by HPLC on a chiral stationary phase. dConversion (C) = ees/(ees+eep). eS factor (S) = ln[(1 – C) (1 – ees)]/ln[(1 – C) (1 + ees)].
为了深入地探究这一有趣现象背后的机理,他们开展了一系列控制实验(Figures 2a & 2b)并对反应体系中E/Z两种异构体的相对组成和对映选择性随时间的变化情况进行了详细记录(Figure 2c)。结果证明 (S,E)-1a和 (R,E)-1a在反应体系中存在不同的敏化效率,这与动力学拆分过程是一致的,因此也排除了该反应经历去外消旋化的反应途径。紫外可见吸收光谱显示,原位生成的CuI-L1配合物和潜在的反应中间体 [(±)-E-1a]-CuI-L1在400 ~ 450 nm范围内表现出明显的吸收增强,并且能够与427 nm蓝色LED灯的发射光谱发生部分重叠。因此,该波长LED灯能够作为最佳光源用于活性中间体的激发。与此相反,456 nm蓝色LED灯的发射光谱几乎不能与之形成有效的重叠,这与该波长LED灯在反应中无法实现动力学拆分是一致的。
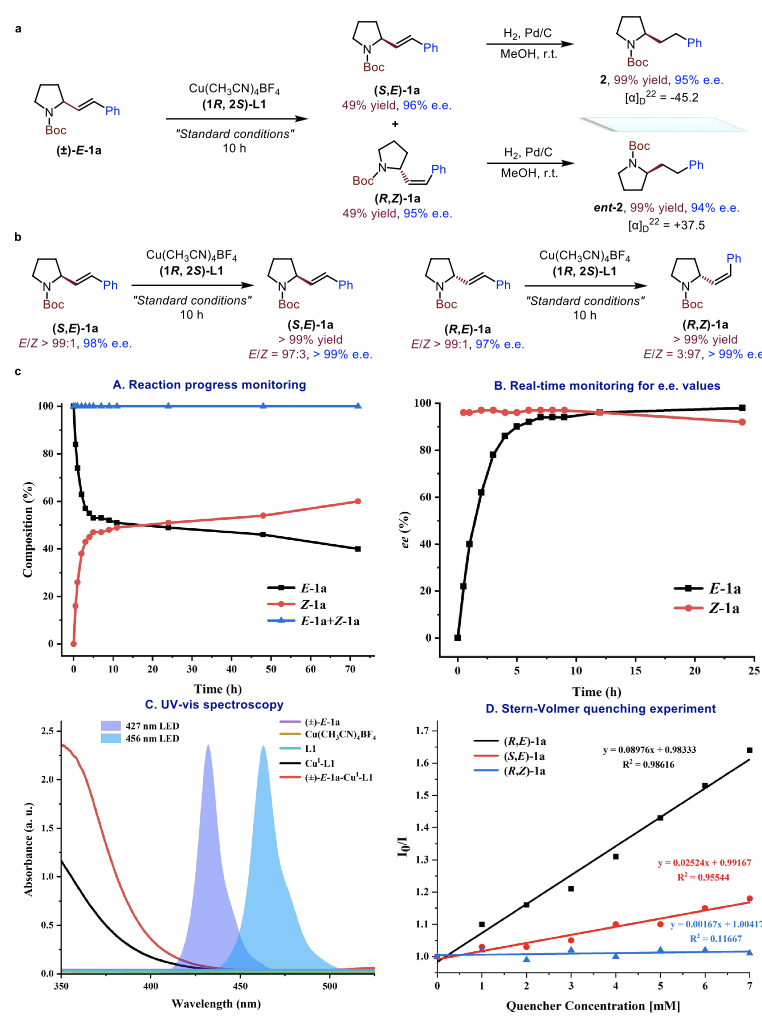
Figure 2. Experimental mechanistic studies of visible-light-induced catalytic kinetic resolution.
随后,作者与曲阜师范大学袁相爱课题组合作,结合机理验证实验以及理论计算对反应机理进行了系统的研究(Figure 3)。理论计算表明,R-构型的吡咯烷底物 (R,E)-1a由于羰基氧原子与铜原子中心的配位作用使其与催化剂形成的复合物在基态时具有更好的稳定性。此外,这种O···Cu相互作用使得R-构型底物在C2−C3单键旋转阶段所克服的电子活化能更低,因此具有更高的敏化效率,最终利用这种总体敏化速率的差异实现高效地动力学拆分过程。
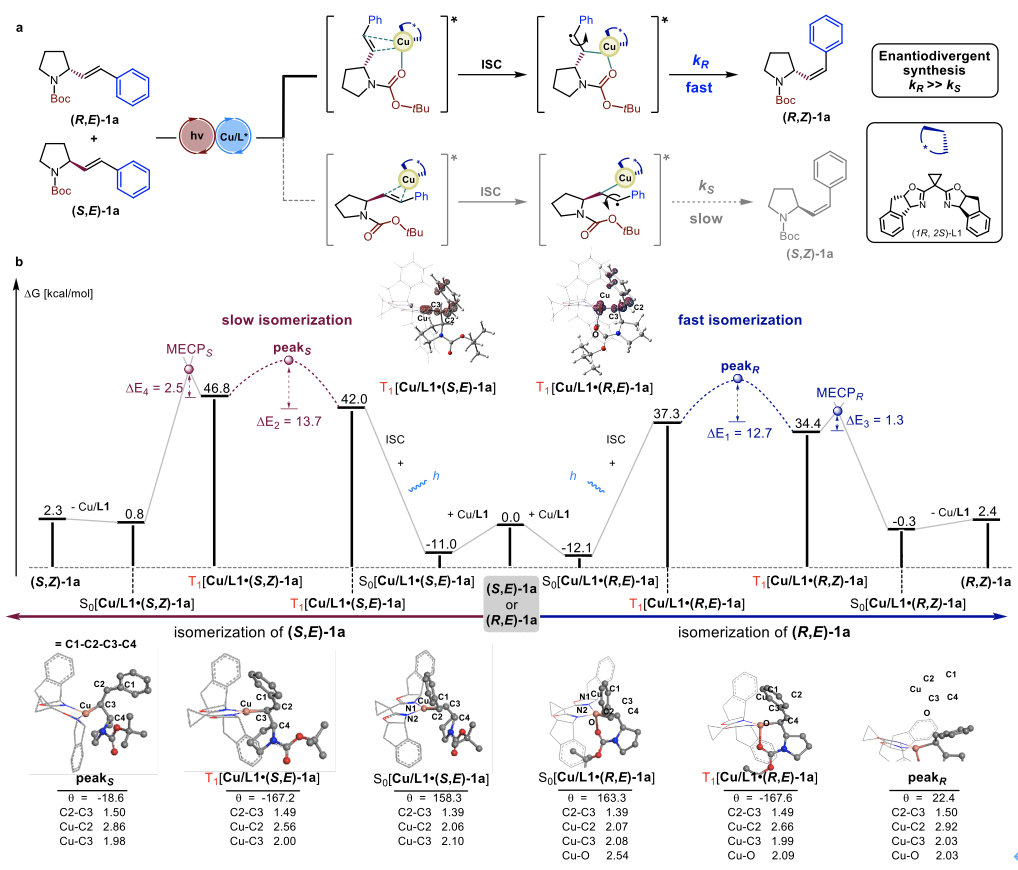
Figure 3. Mechanistic description of photocatalytic kinetic resolution on the basis of DFT calculations.
基于已确定的最佳反应条件,他们对此动力学拆分的底物适用性进行了考察。常见的N-保护基如Boc、Cbz、Bz和Ac在最佳反应条件下都可以很好地兼容。随后,他们使用Boc作为最优的N-保护基,对吡咯烷芳环片段上的修饰进行了考察。芳环的电子效应对动力学拆分效率影响甚微,三氟甲基、卤素、羧酸酯和硼酸酯等官能团都可以很好地兼容。最后,他们尝试对吡咯烷骨架进行修饰,结果发现吡咯烷骨架C4-位氟原子和TBS保护羟基的引入对动力学拆分效果影响不大,均能够顺利地以较高对映选择性得到所需的异构化产物。
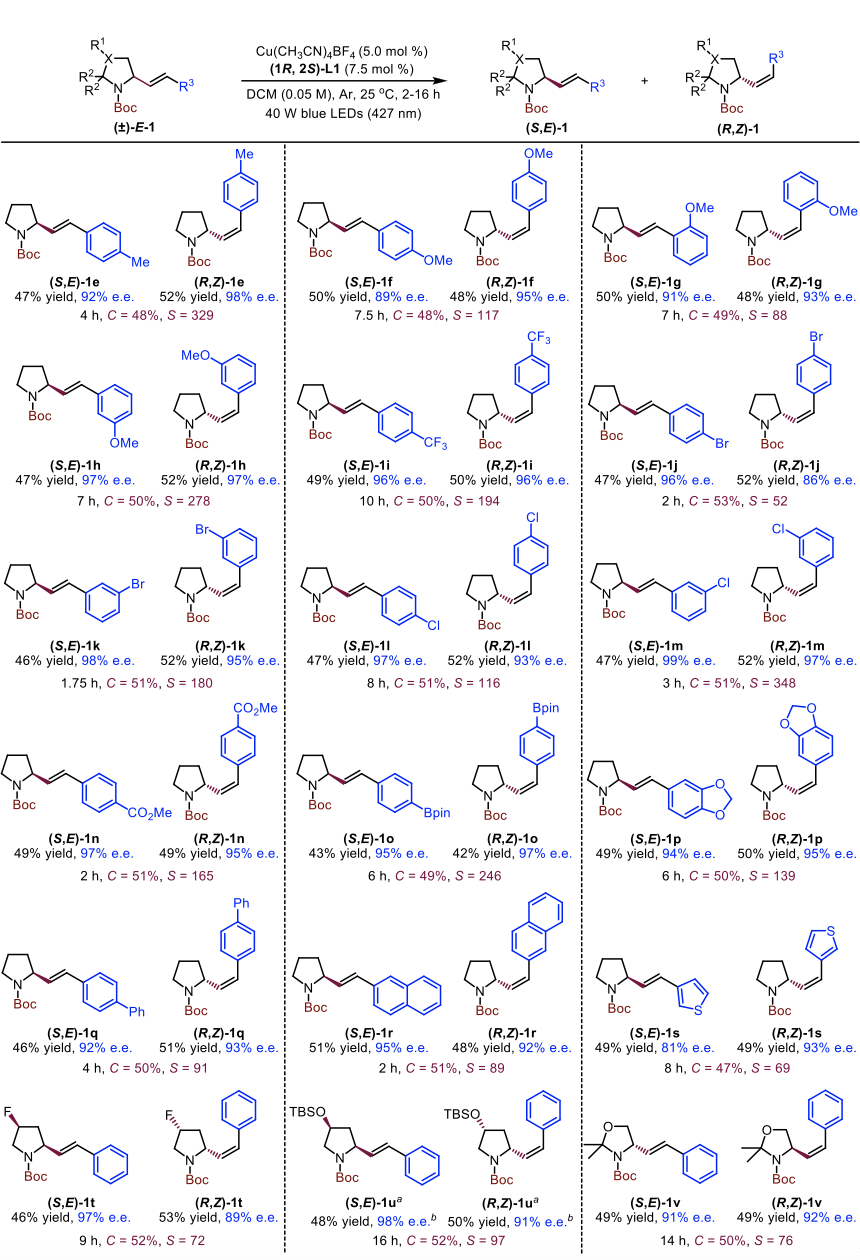
Figure 5. Substrate scope for kinetic resolution of racemic N-Boc-2-styrylpyrrolidines derivatives.
为了验证该动力学拆分策略在有机合成中的实用性,他们使用同一手性配体的一对对映异构体成功实现了四种立体异构体[(R,Z), (S,Z), (S,E)和(R,E)]的立体发散性合成(Figure 6a)。为了进一步扩大该策略的合成应用价值,他们在最佳反应条件下对(±)-E-1m 进行了克级规模(3.5 mmol)的放大尝试。该动力学拆分反应的效率并不会因为反应量的扩大而显著降低。最后,他们利用 (S,E)-1m 和 (R,Z)-1m作为起始原料,分别实现了生物活性分子indolizidine生物碱201两种对映体的全合成(Figure 6b)。
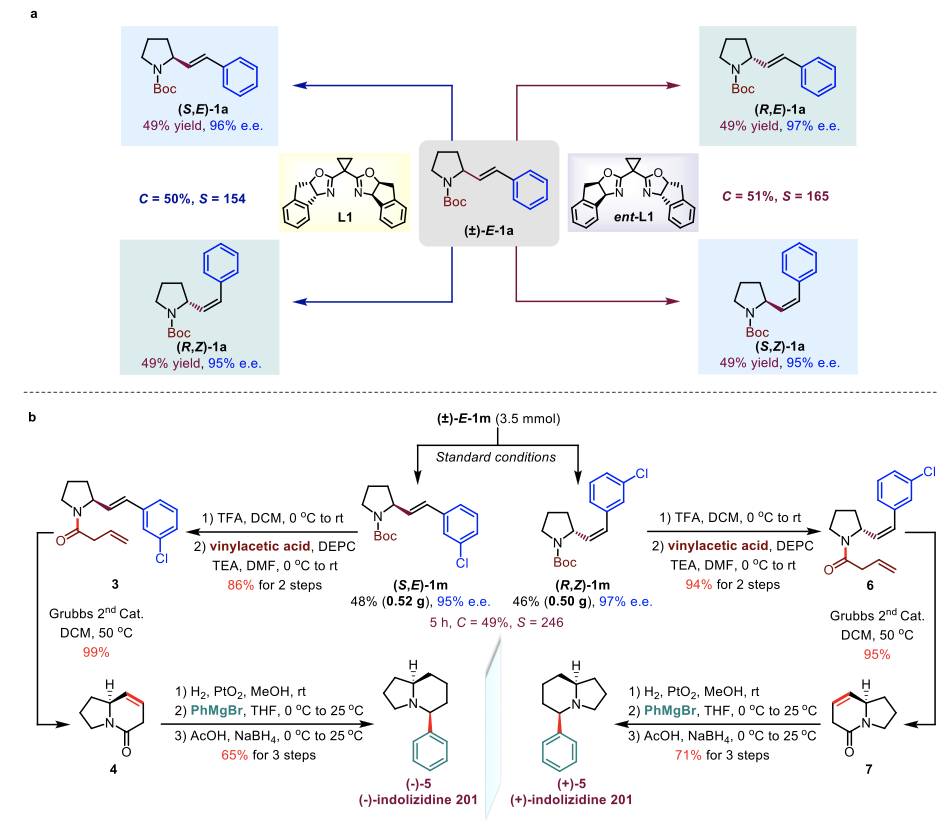
Figure 6. Stereodivergent synthesis of 2-styrylpyrrolidine 1a (a) and total synthesis of indolizidine 201 (b).
总结
南京大学俞寿云课题组与曲阜师范大学袁相爱团队合作,发展了一种基于光激发手性铜配合物介导的对映选择性E → Z异构化策略,成功实现了一系列外消旋2-苯乙烯基吡咯烷衍生物的动力学拆分。机理验证实验和DFT计算表明,底物−催化剂复合物的两个对映体总体敏化速率的不同,是该异构化过程具有优异动力学拆分效率的决定性因素。该方法进一步拓展了动力学拆分的基态反应模式,为激发态对映选择性催化过程提供一个新的思路。南京大学张昊博士为论文第一作者,俞寿云教授和袁相爱副教授为共同通讯作者。
参考文献
- [1] J. B. Metternich, R. Gilmour, J. Am. Chem. Soc. 2015, 137, 11254. doi: 10.1021/jacs.5b07136.
- [2] K. Singh, S. J. Staig, J. D. Weaver, J. Am. Chem. Soc. 2014, 136, 5275. doi: 10.1021/ja5019749.
- [3] (a) H. Zhang, Q. Xu, L. Yu, S. Yu, Eur. J. Org. Chem. 2020, 2020, 1472. doi: 10.1002/ejoc.201901119.
- (b) X. Shen, C. Huang, X. Yuan, S. Yu, Angew. Chem. Int. Ed. 2021, 60, 9672. doi: 10.1002/anie.202016941.
- [4] (a) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer, F. Glorius, Chem. Soc. Rev. 2018, 47, 7190. doi: 10.1039/C8CS00054A.
- (b) Q. Zhou, Y. Zou, L. L u, W. Xiao, Angew. Chem. Int. Ed. 2019, 58, 1586. doi: 10.1002/anie.201803102.
- (c) F. Strieth-Kalthoff, F. Glorius, Chem. 2020, 6, 1888. doi: 10.1016/j.chempr.2020.07.010.
- [5] A. Hossain, A. Bhattacharyya, O. Reiser, Science 2019, 364, eaav9713. doi: 10.1126/science.aav9713.
- [6] X. Huang, E. Meggers, Acc. Chem. Res. 2019, 52, 833. doi: 10.1021/acs.accounts.9b00028.
- [7] G. E. M. Crisenza, A. Faraone, E. Gandolfo, D. Mazzarella, P. Melchiorre, Nat. Chem. 2021, 13, 575. doi: 10.1038/s41557-021-00683-5.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载




No comments yet.