
本文作者:孙苏赟
第四部分 软亲核试剂的反应
1. 软亲核试剂与内消旋化合物和1,1-位同取代基的烯丙基化合物的反应
(1) 内消旋化合物
常见的内消旋烯丙基化合物有以下三类,这些烯丙基化合物发生过渡金属的氧化加成产生一对对映异构体。
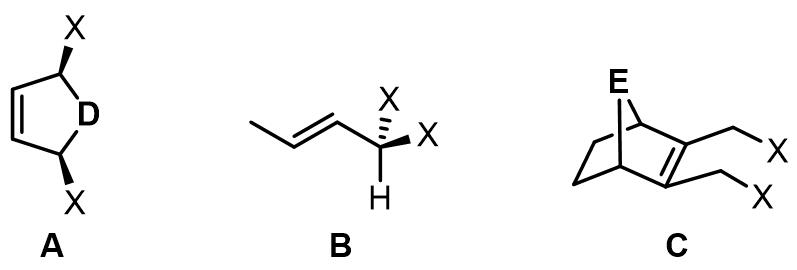
A类化合物发生氧化加成时,过渡金属和另外一个离去基团处于烯丙基平面的异侧,此使在那一侧发生烯丙基配位直接影响反应最终的立体化学。B类化合物是偕取代的,过渡金属将哪个X基团离子化是立体化学的关键[1,2]。C类化合物的研究却不很多,主要在一些胺化反应中使用。
目前,对于A和B类内消旋底物的不对称Tsuji-Trost取代反应的研究较多。对于环状A类底物,常用的配体是邻(二苯基膦)苯甲酸(diphenylphosphino benzoic acid, DPPBA)类配体[3]。
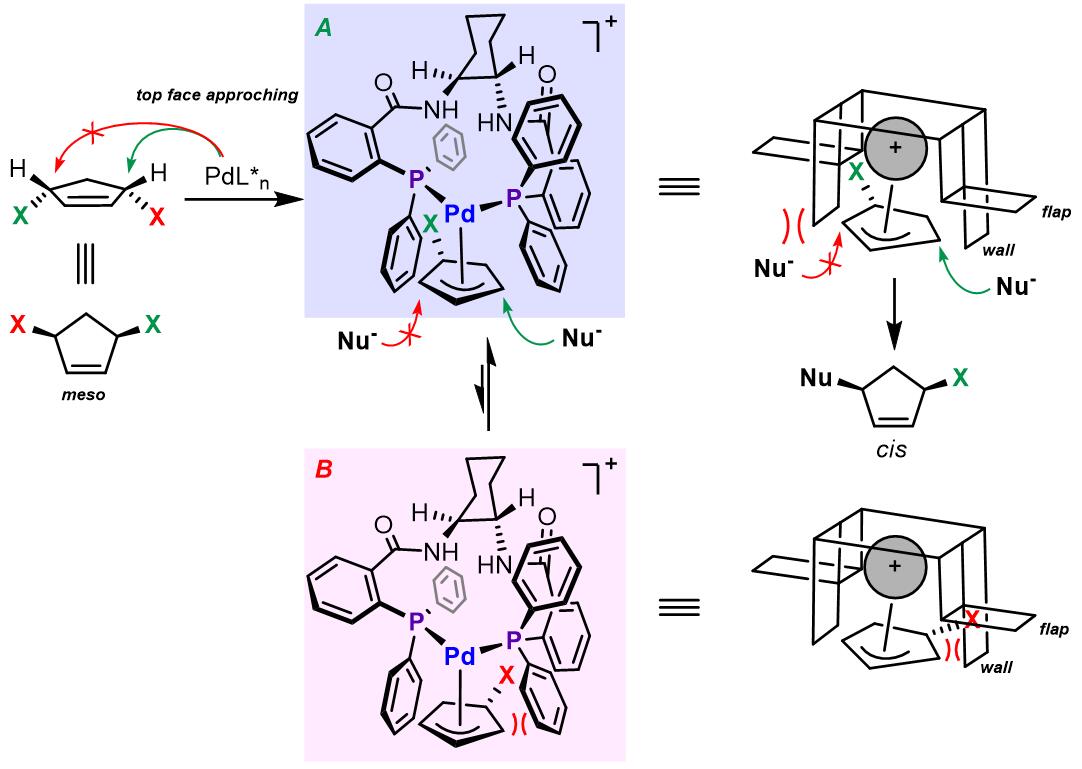
这类配体的工作原理可以用一个简易的模型进行解释。在烯丙基配合物形成步骤,配体也可对离去基团离子化进行区域选择性产生影响,如果烯丙基和Pd进行配合得到配合物B,烯丙基结构上的另一个离去基团回和配体上的苯基发生因位阻产生的重叠,因此烯丙基配合物更倾向于以A配合物的形式存在。在这,又因为配体中四个苯基的屏障和襟翼的作用使得亲核试剂可以高度选择的发生在位阻更小的位点最终得到cis-产物。
此类反应主要以1,4-二醇衍生物的反应为主,这里是几个例子:
(a)[9]
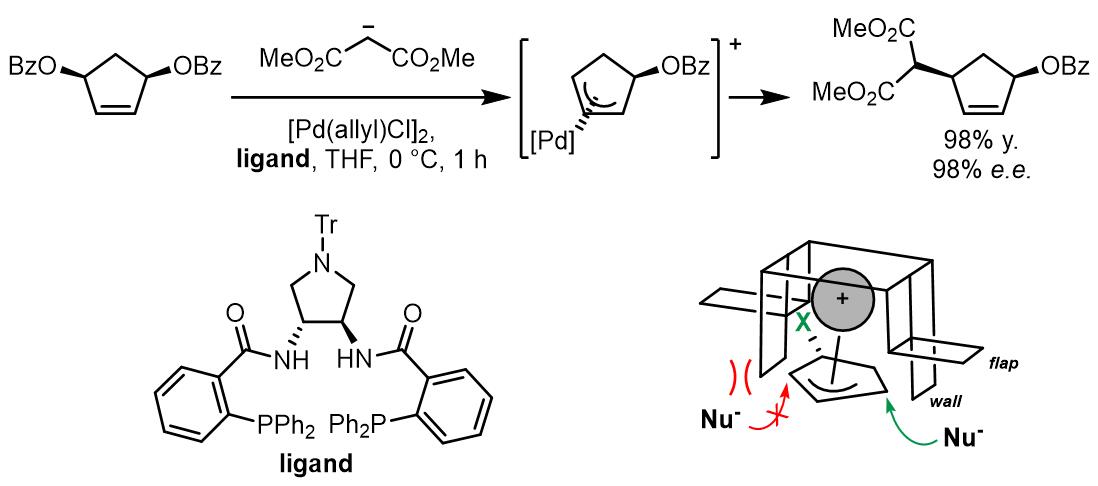
(b)在腺苷的合成中的应用[4]
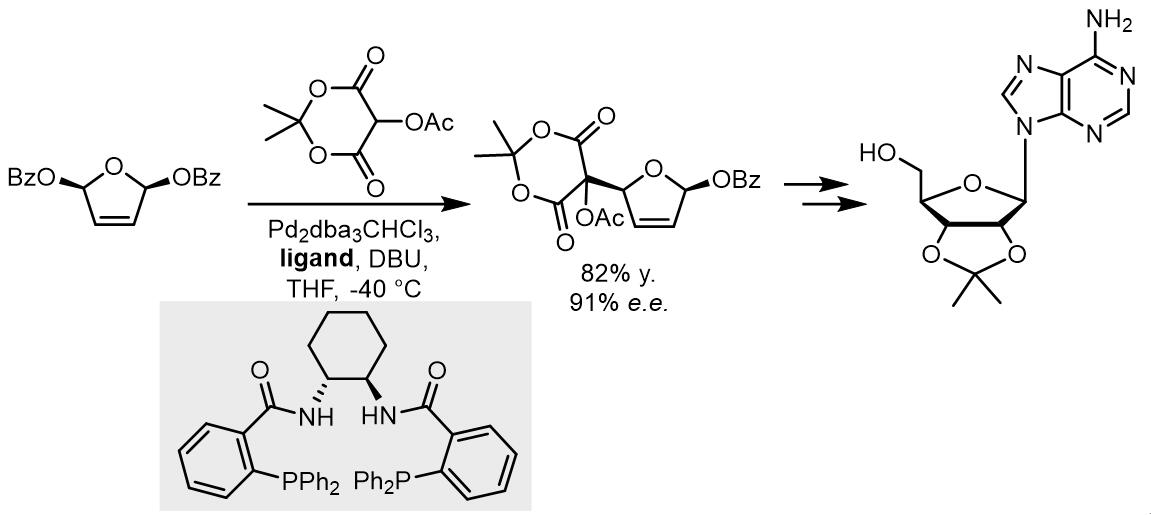
(c) (-)-carbovir的合成[5]

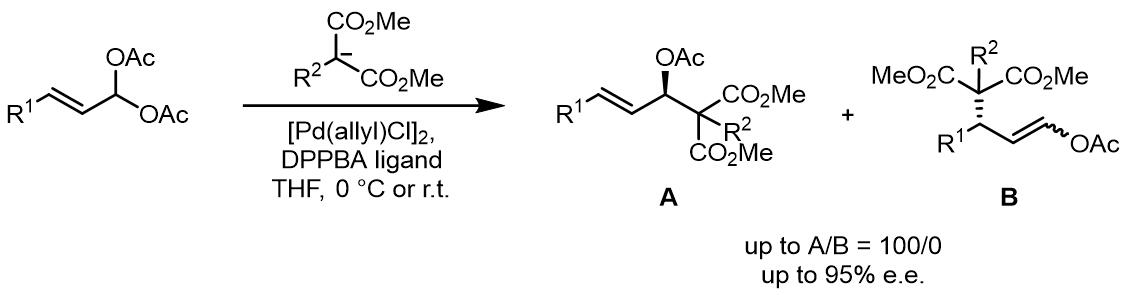
(2) 1,1-位同取代基的烯丙基化合物
以烯丙基1,1 -偕二醇羧酸酯为例,在合成上他们和不对称亲核加成的醛化合物是等价的有机合成砌块。值得注意的是,反应的区域选择性遵循普遍规则,端位的O-取代基会使得亲核试剂倾向于在取代基的近端进攻。
类似的,此类化合物的不对称Tsuji-Trost取代反应常用的配体还是DPPBA类配体,但是产物的ee值不如环状内消旋化合物的反应的产物[6,7]。
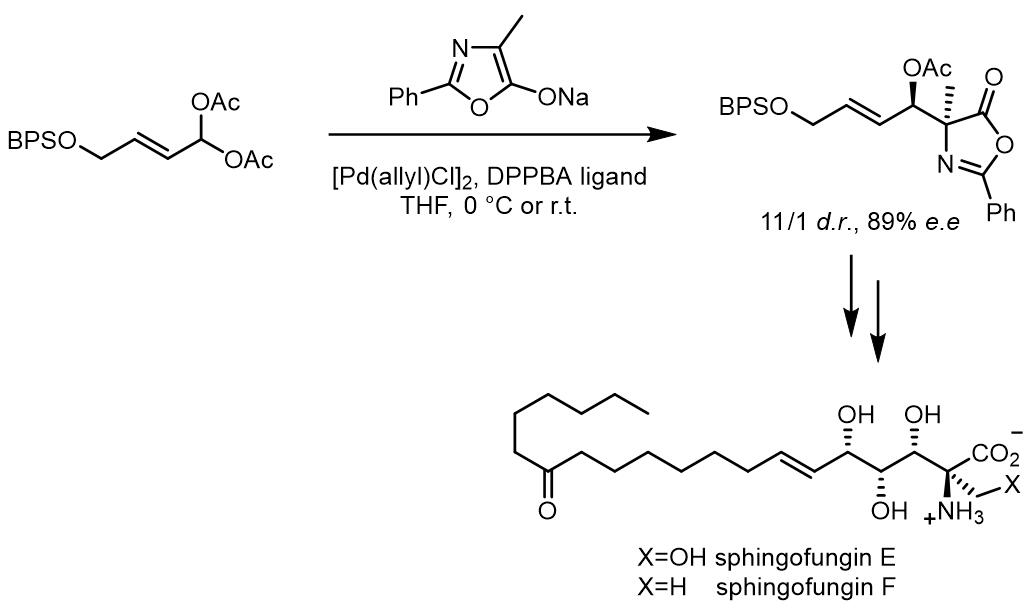
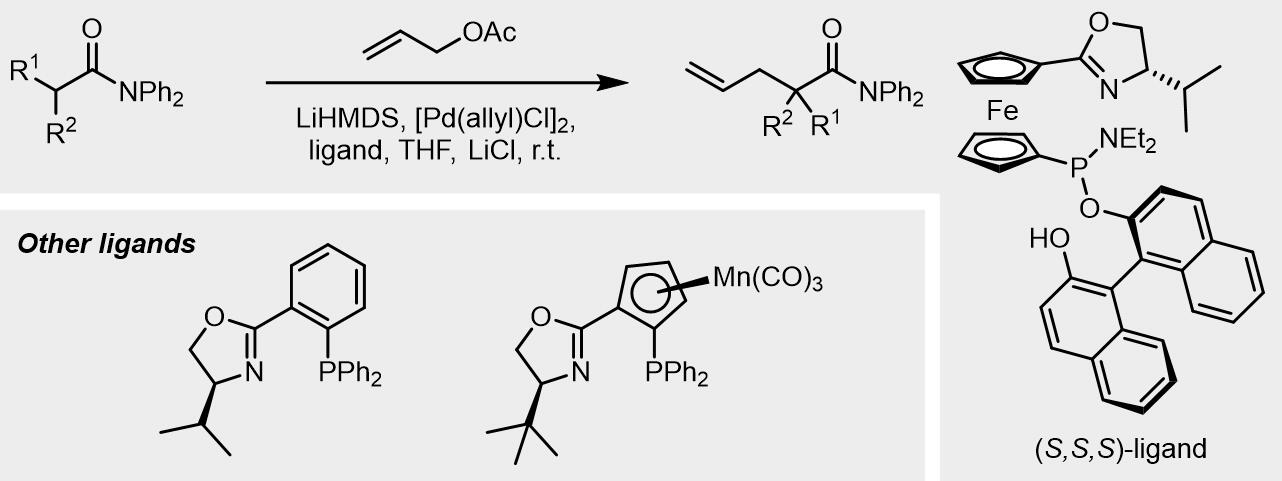
除了丙二酸碳负离子类亲核试剂,噁唑烷酮类衍生物可作为亲核试剂,得到的产物具有α-氨基酸结构,因此在天然产物的合成中有优势[8]:
2. 软亲核试剂和1,3-位置相同取代基的烯丙基配合物的反应
这类1,3-位置相同取代基的烯丙基配合物是在过渡金属氧化加成后得到的烯丙基-Pd配合物是对称的,主要由以下两种类型:
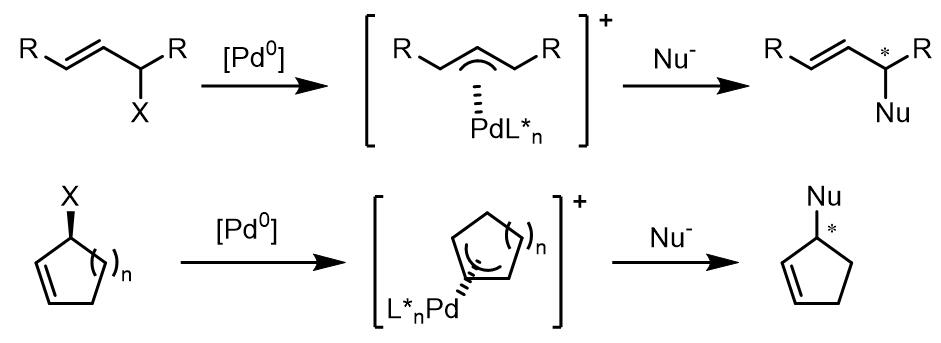
若需要实现这类反应的不对称加成,除了使用经典的DPPBA类配体,还有另外开发了几类手性配体。
(a)DPPBA类配体
DPPBA类配体的立体化学选择性上最重要的是P-Pd-P键角,键角越大则烯丙基-Pd结构越暴露在配体以外,反之烯丙基-Pd暴露越少而立体化学选择性相对更好。
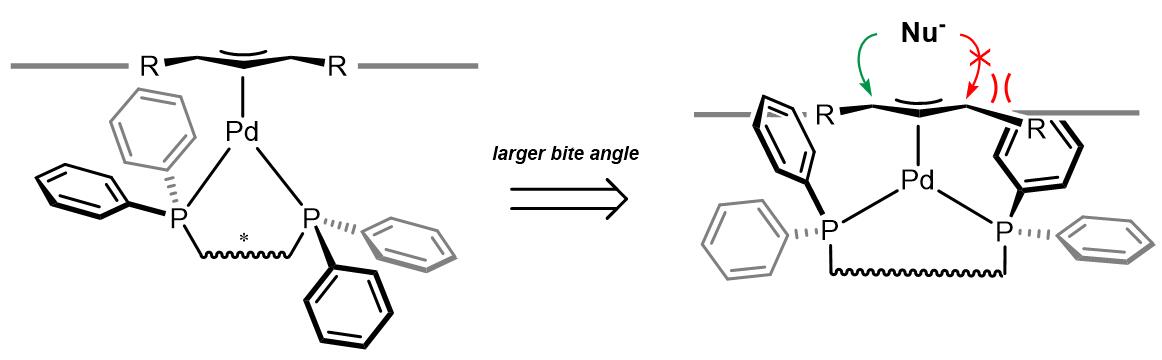
(b)C2-轴对称类配体
Pfaltz开发出BOX类配体,这类配体也可以用于不对称 Tsuji-Trost取代反应。例如R=Bn时[10]:
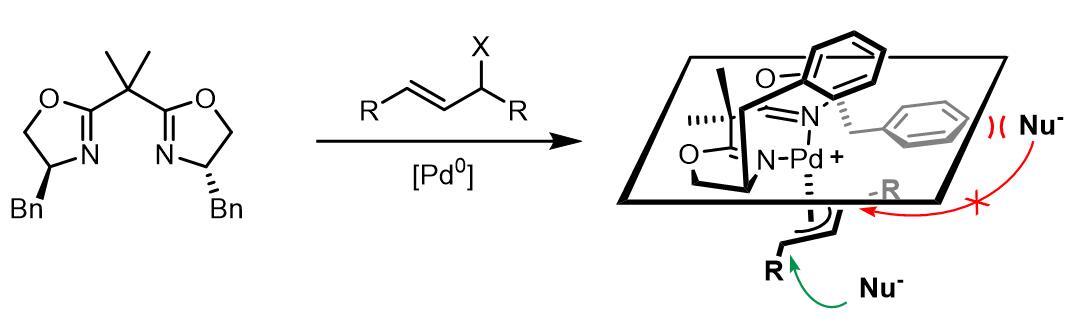
(这里需要注意:BOX配体在上述配合物中不是完全平面结构,以上只是示意图)
(c) 导向基团类配体[11]
利用羟基和亲核试剂之间的氢键作用可以使得设计出一类二茂铁催化剂配体以完成高度选择性的烯丙基取代反应:
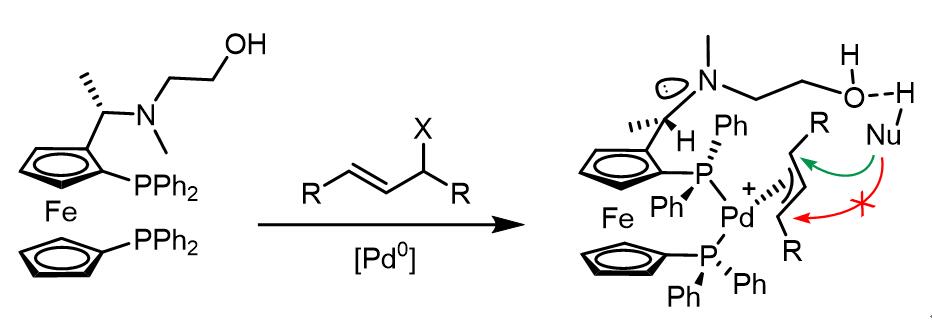
(d) 电子密度差异化配体
此类配体主要有PHOX和QUINAP类等配体:
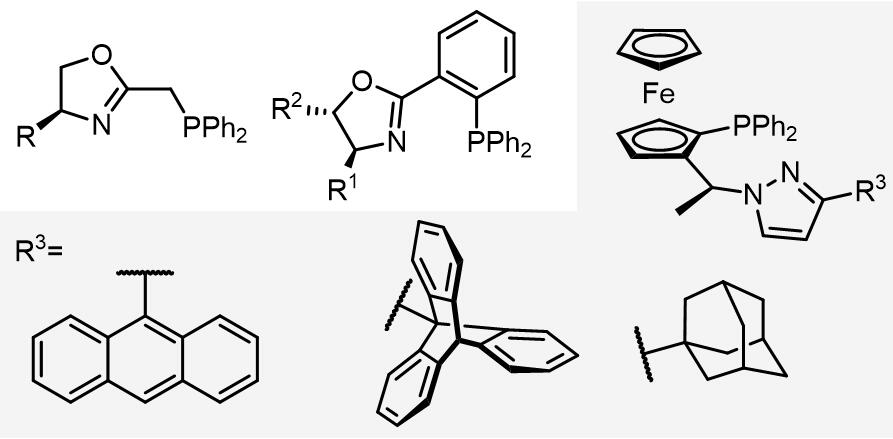
(e) 结构不对称的配体[12-18]
若使用不对称的配体,生成的烯丙基配合物可以在endo-和exo-配合物快速异构化,理论上会有四种不同的反应途径,但是此类配体可以得到高度立体专一的不对称取代反应的产物。
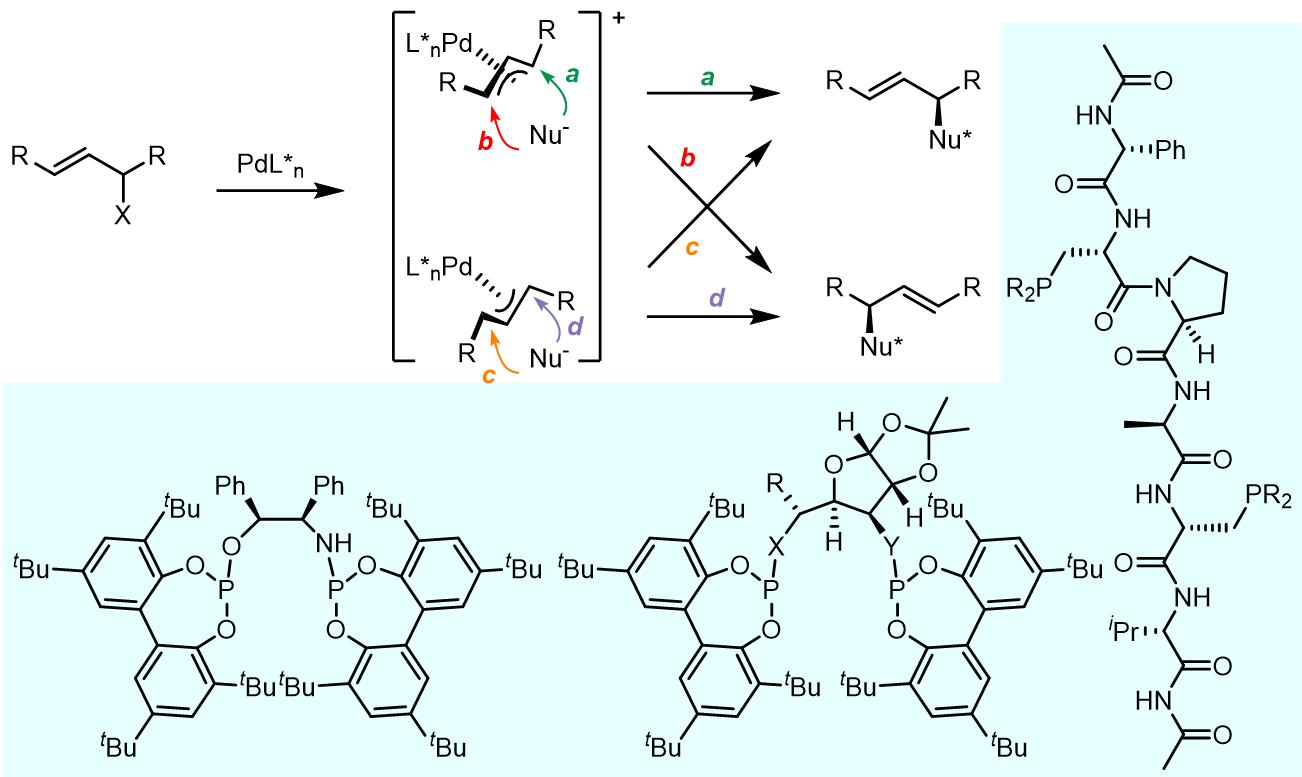
3. 亲核试剂和单取代和1,1-偕二取代的烯丙基配合物的反应
当烯丙基上两端碳原子上的两个取代基相同,那么得到的烯丙基配合物具有手性,但是两种烯丙基配合物(一对对映异构体)之间存在平衡,但是专一的得到一种立体化学的产物需要通过配体进行调控。
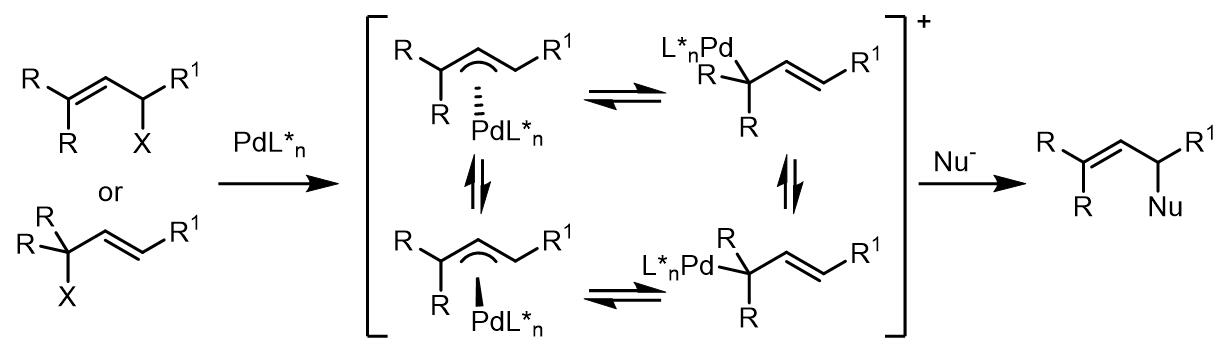
通常来讲,反应的区域选择性可以通过分子内烯丙基Tsuji-Trost取代反应来实现,也就是说环的大小可以对亲核试剂进行导向作用使得进攻位阻较小的位置。但是目前为止Pd催化体系的分子间反应的结果并不是很好。

对于这类反应,可能会有四种不同的过渡态,SN1类的过程在枝状化合物的反应中可以得到高立体选择性的,如果使用的配体供电性较弱或是吸电子配体,效果会更好[19]。此外,配体的位阻效应可以对烯丙基配合物的结构进行选择,并且对亲核试剂的进攻位点进行控制(C和D)[20],效果最好的配体还是G配体。使用H配体的时候,配体和过渡金属的化学计量比不会影响反应的区域选择性,反应的过程也十分详细:
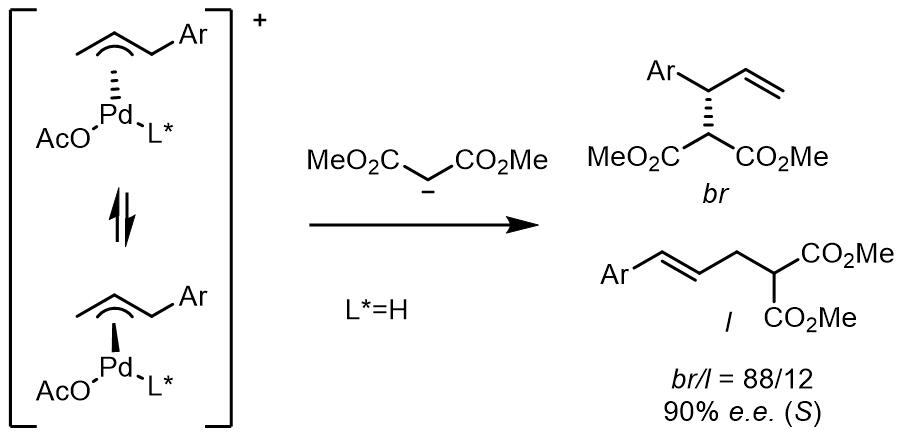
戴立信院士和侯雪龙研究员分别对这个问题开发出一些解决方案(配体F),使用P,N-双齿配体可以造成电子分布的不均匀,此外BINAP类似的配体也可以起到类似的作用。
三取代烯丙基配合物的反应,传统的DPPBA类、sparteine配体催化也是适用的,即使底物是联烯醇类衍生物,立体化学选择性的效果也普遍不错:
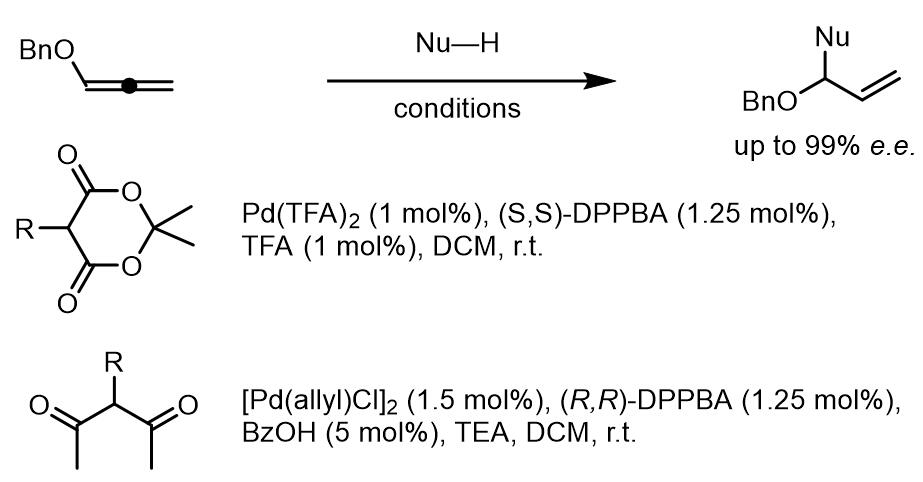
另外,PHOX类大位阻配体也很适用于此类反应[21-23]:
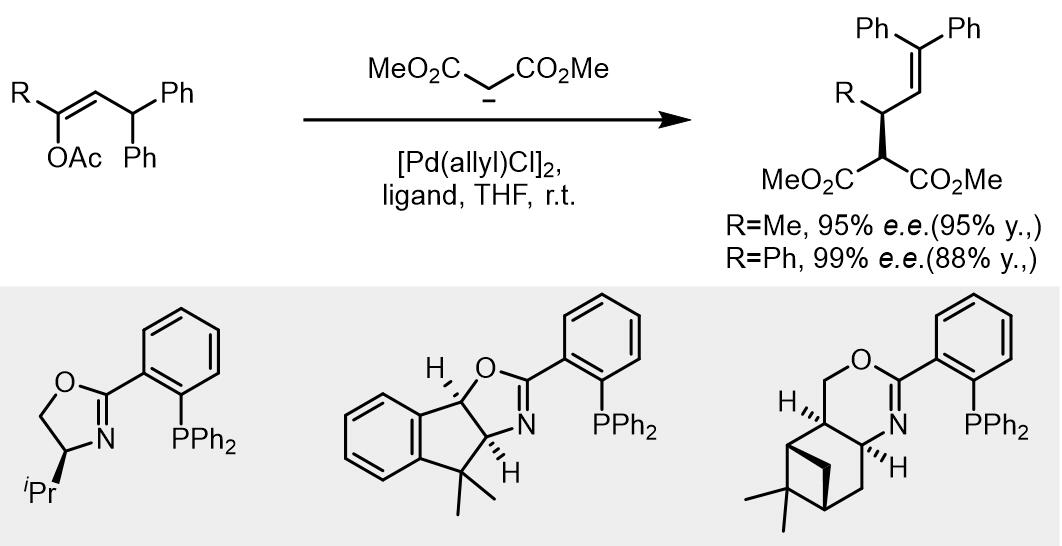
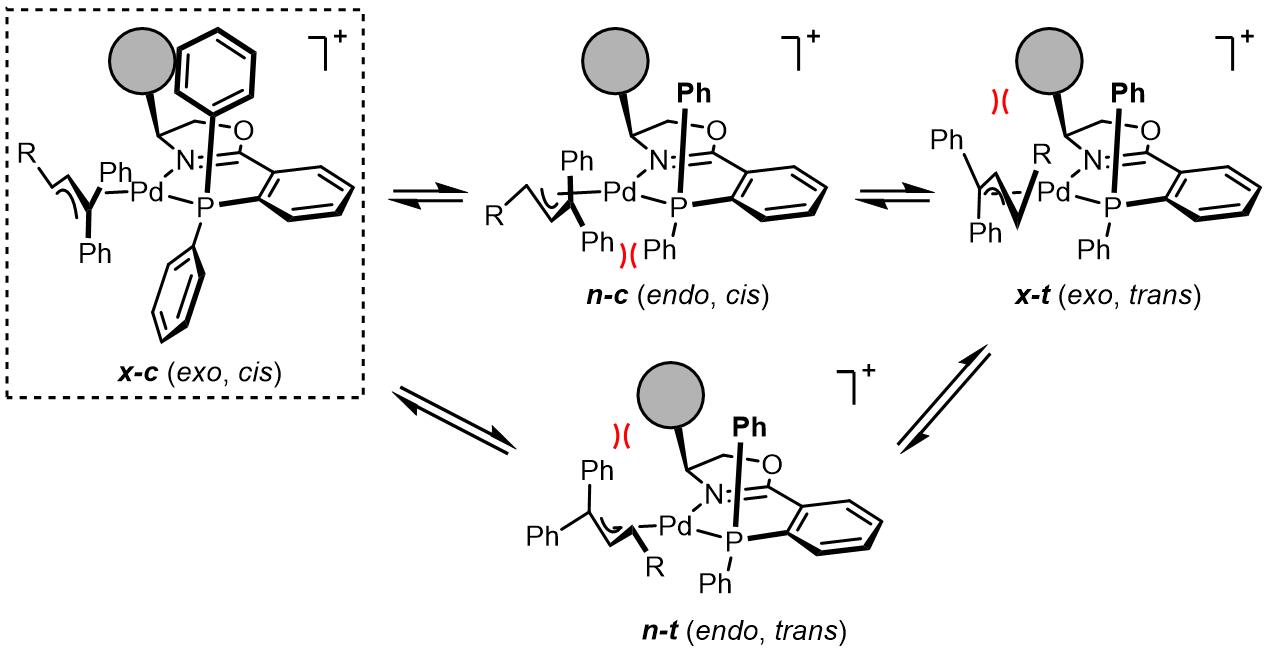
反应的烯丙基配合物因为exo/endo (x/n)和cis/trans (c/t)的组合会有四种不同可相互转化的构型,因为烯丙基上取代基和配体上大位阻基团的相互作用而只有x-c结构最稳定。
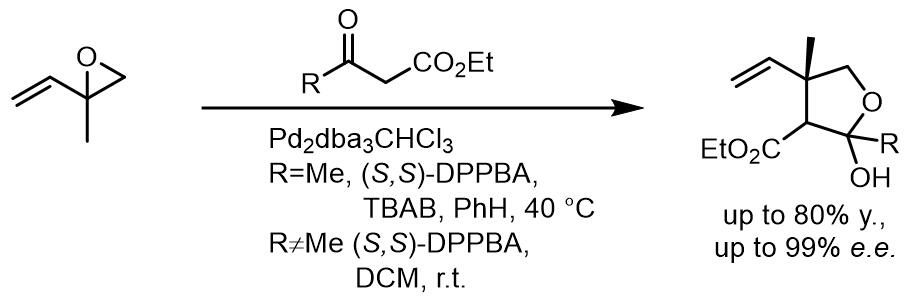
一类特殊的底物是烯丙基环氧化物[24],环氧化合物的活性都很高,通常得到的是线性化合物,在氧化加成后的烯丙基配合物中氧原子会对亲核试剂由导向作用。对于DPPBA类配体,由于空间上的限制作用,反应的效果会很好。

这里是一个应用[25]:
4. 由烯丙基化合物制备联烯
从烯丙基类化合物到联烯的转化在天然产物的合成中使用较多,目前有两种比较主流的方式,分别是从联烯底物和共轭丁二烯底物。氧化加成后得到的烯丙基配合物可以和软亲核试剂在位阻较小的碳原子上发生取代反应, 并且此类反应可以用于发展动力学拆分反应。
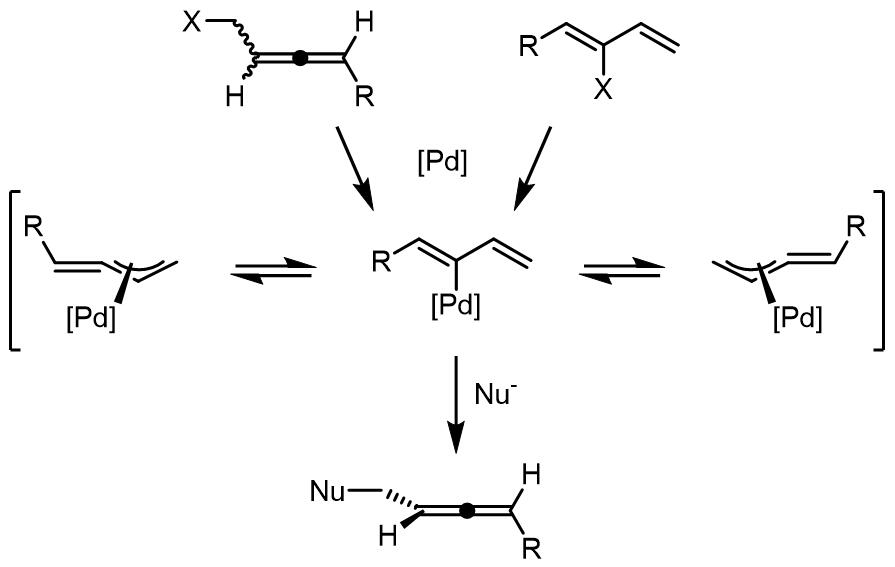
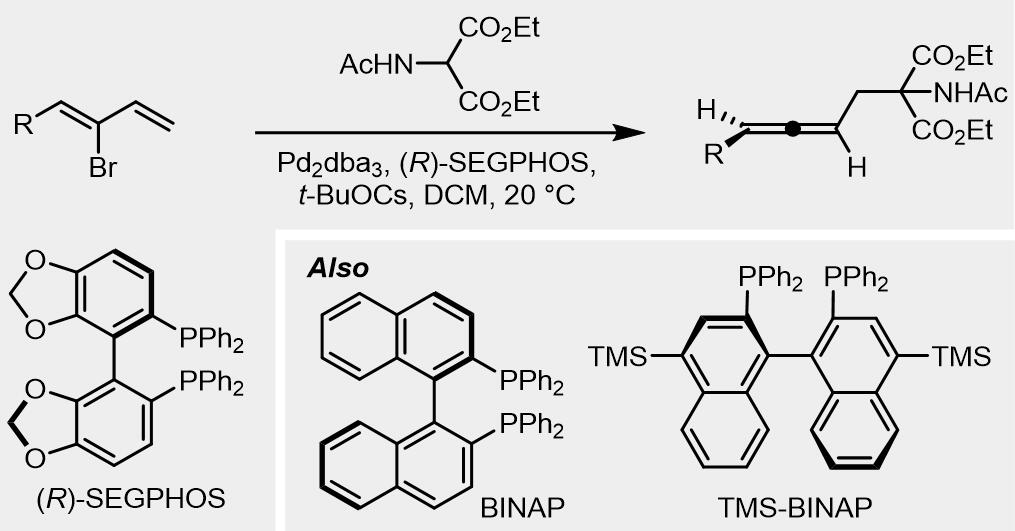
对于1-取代的底物使用Br作为离去基团,反应使用的配体主要是BINAP类[26],之后还开发出了TMS-BINAP,SEGPHOS作为配体也有不错的效果[27,28]:
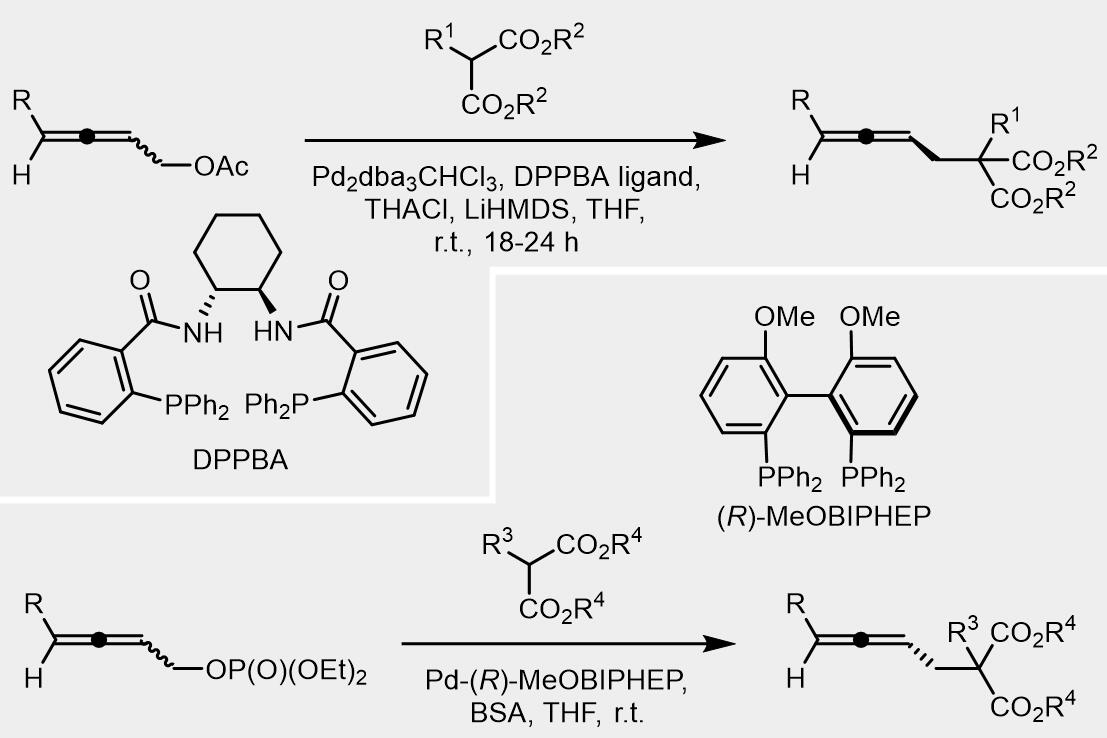
而对于联烯底物,常使用磷酸酯和醋酸酯作为离去基团:
5. 潜手性亲核试剂
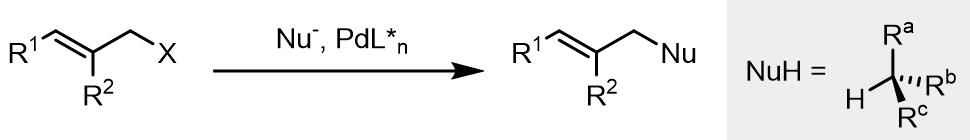
此类反应由上世纪70年代开始开发,立体选择性与之前的原理类似,主要有以下几类配体-过渡金属体系以适用于不同的潜手性亲核试剂:
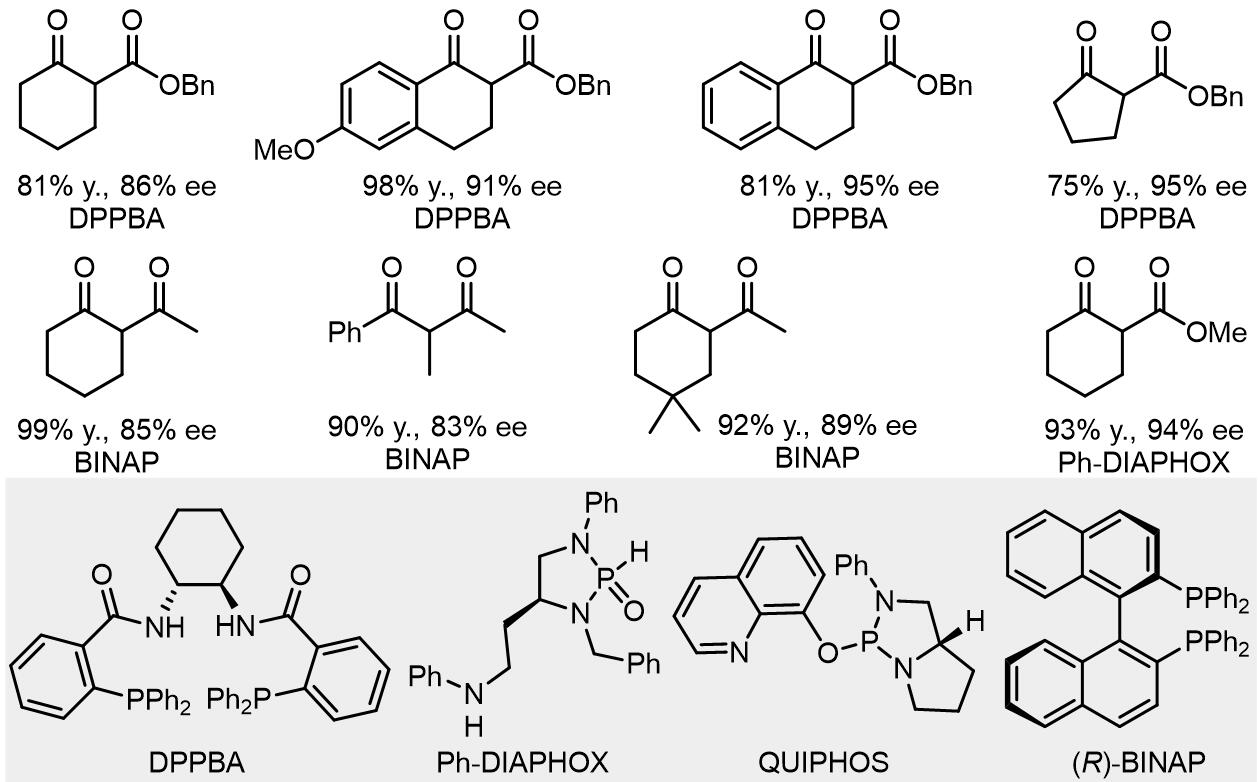
(1) β-二羰基化合物
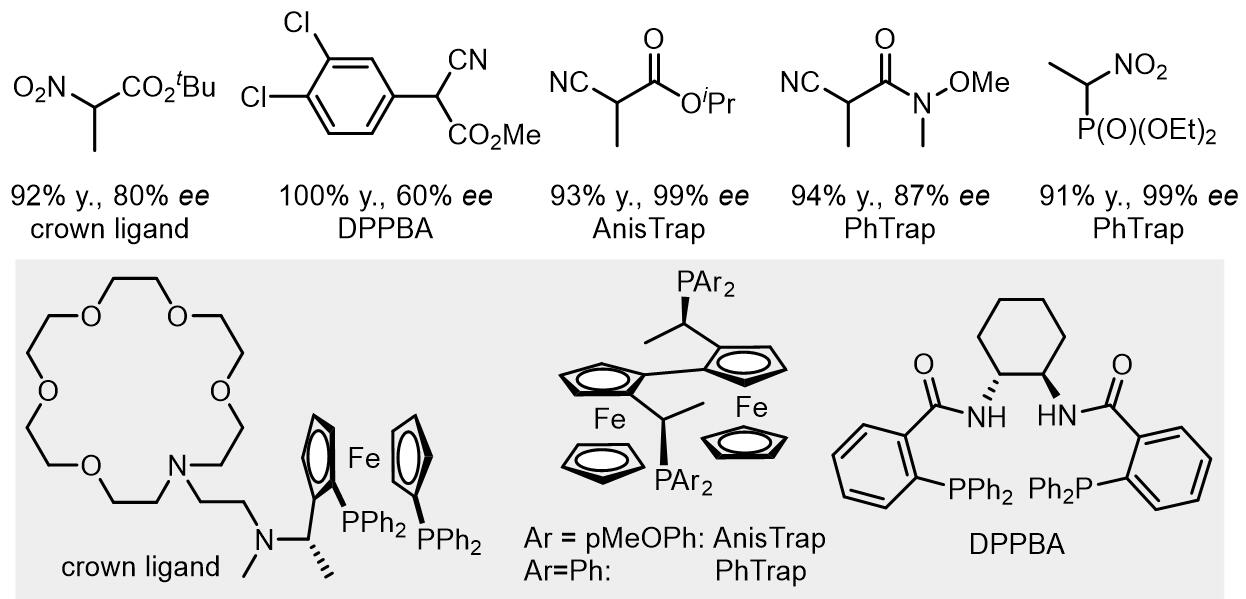
(2) α-氰基羧酸衍生物
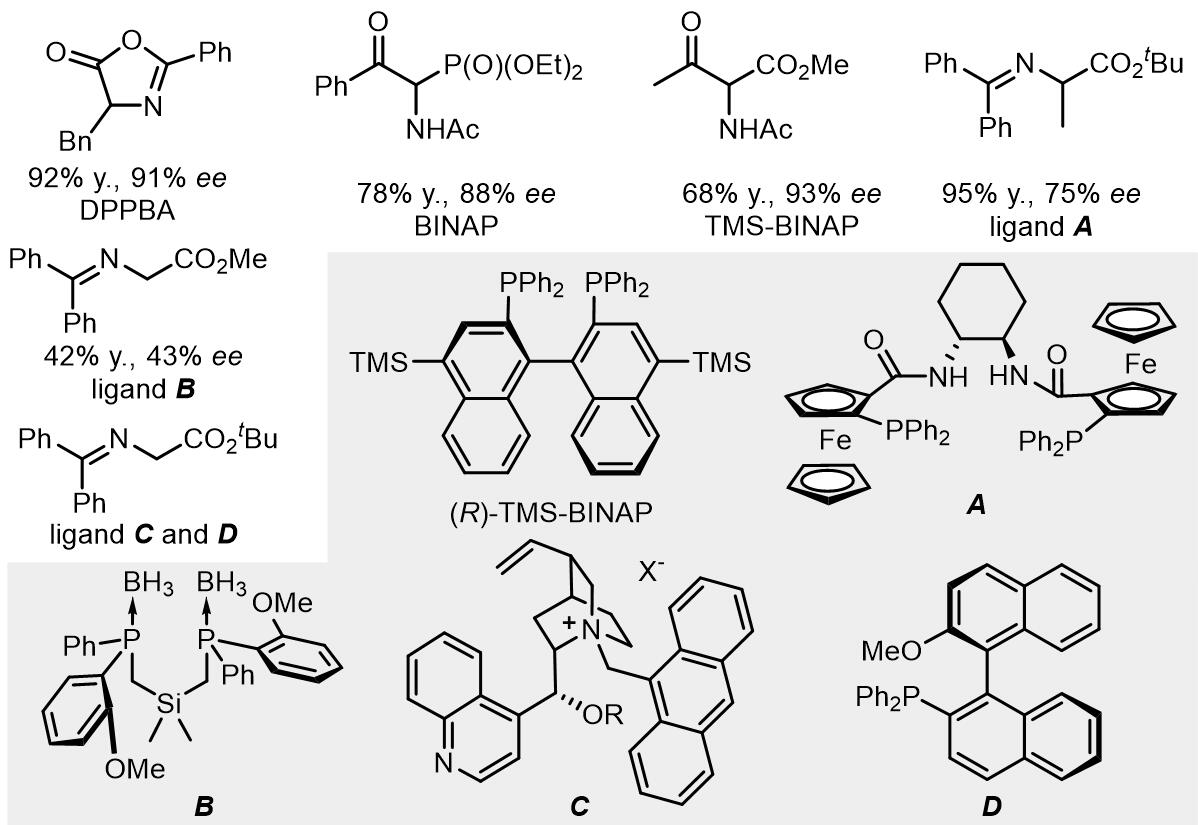
(3) 氮杂内酯化合物,α-氨基酮,α-氨基酸衍生物
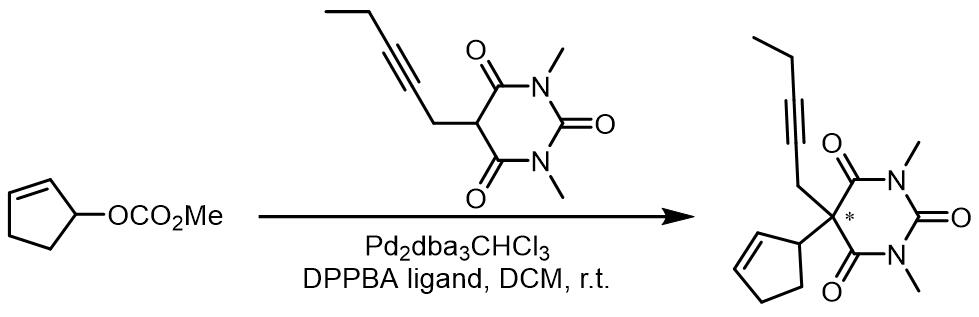
(4) 巴比妥酸其衍生物
巴比妥酸衍生物的例子目前不是很多,这里是一个例子:
6. 烯醇衍生物亲核试剂
对于金属试剂作为亲核试剂的不对称烯丙基烷基化,例如有机锌试剂和有机镁试剂,Pd催化的效果一般不太好,但是酮和酯的烯醇负离子的反应可以做到较好的立体化学选择性。往往这类反应会生成两个手性中心,而亲核试剂片段上的手性比较难以控制。
(1) 酮烯醇试剂
1980年,Trost和Keinan报道了一个酮烯醇负离子作为亲核试剂的反应,但是收率并不是太好[29],之后Fiaud和Malleron开发出了利用体积更大的烯醇负离子亲核试剂[30]。在1999年,Trost和Schroeder报道了首例钯催化烯醇锡试剂的不对称烯丙基烷基化反应[31,32],相似地,反应中使用的是DPPBA类配体,达到了高收率和高度的立体化学选择性,原理和之前的反应也是相同的。
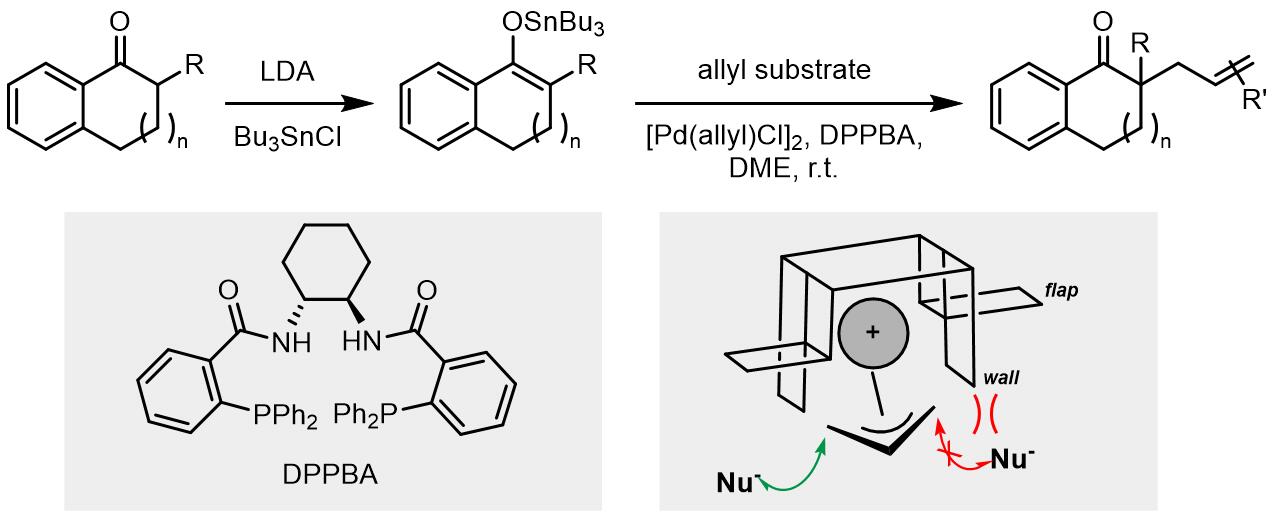
烯醇硅试剂常用作烯丙基醋酸酯的烷基化过程[33,34]:
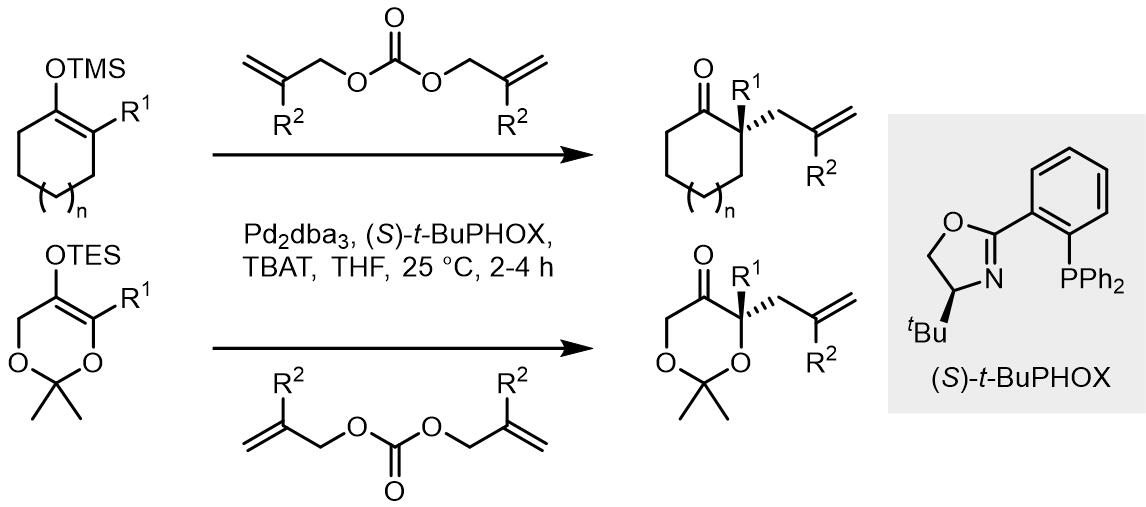
α-氟酮中的氟原子也可以受到影响[35]:
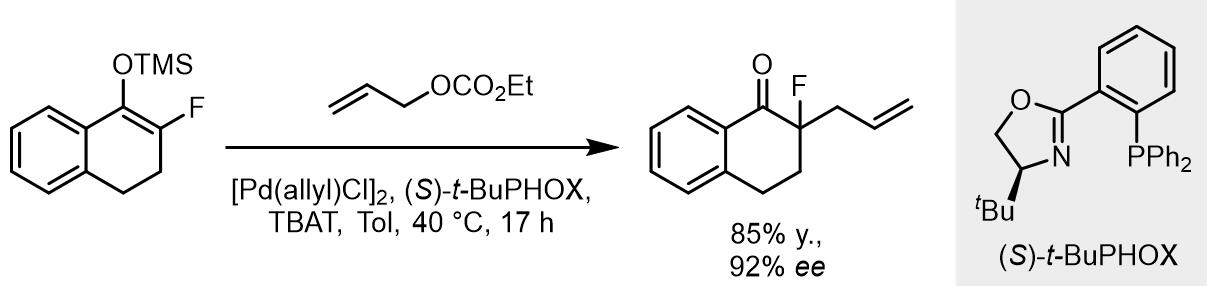
烯醇镁试剂的反应:
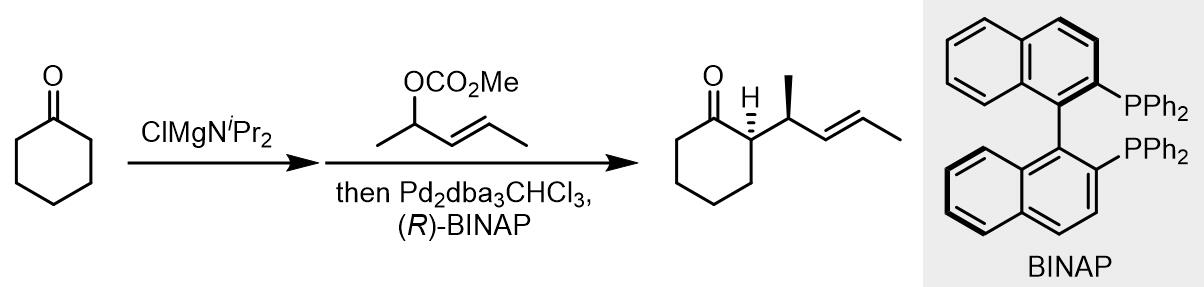
(2) 酯和酰胺烯醇试剂
烯醇作为亲核试剂的反应使用的配体开发较多,这里是一个比较有代表性的反应[36]:
References
- [1] J. Am. Chem. Soc. 2007, 129, 23, 7242–7243. DOI: 10.1021/ja0715896
- [2] Org. Lett. 2006, 8, 3, 519–522. DOI: 10.1021/ol052859x
- [3] Acc. Chem. Res. 2006, 39, 747-760. DOI: 10.1021/ar040063c
- [4] Org. Lett. 2003, 5, 7, 1143–1146. DOI: 10.1021/ol0300219
- [5] Org. Lett. 2003, 5, 7, 1143–1146. DOI: 10.1021/ol0300219
- [6] J. Am. Chem. Soc. 2001 , 123 , 3671 – 3686 . DOI: 10.1021/ja003774o
- [7] J. Am. Chem. Soc. 2001 , 123 , 3687 – 3696 . DOI: 10.1021/ja003775g
- [8] Tetrahedron 1992 , 48 , 2143 . DOI: 1016/S0040-4020(01)88880-3
- [9] J. Am. Chem. Soc. 2006 , 128 , 10674 – 10675. DOI: 10.1021/ja0637954
- [10] Acc. Chem. Res. 1993, 26, 6, 339–345. DOI: 10.1021/ar00030a007
- [11] J. Am. Chem. Soc. 1989, 111, 6301-6311. DOI: 10.1021/ja00198a048
- [12] Chem. Commun., 2001, 1132-1133. DOI: 10.1039/B101567M
- [13] J. Org. Chem. 2001, 66, 26, 8867–8871. DOI: 10.1021/jo0159284
- [14] Adv. Synth. Catal. 2005 , 347 , 1257 – 1266. DOI:10.1002/adsc.200505013
- [15] J. Org. Chem. 2005, 70, 9, 3363–3368. DOI: 10.1021/jo0480904
- [16] Org. Lett. 2007, 9, 1, 49–52. DOI: 10.1021/ol0624631
- [17] Chem. Eur. J. 2008 , 14 , 944 – 960. DOI: 10.1002/chem.200700852
- [18] J. Org. Chem. 2007, 72, 8, 2842–2850. DOI: 10.1021/jo062311j
- [19] Angew. Chem. Int. Ed. Engl. 1998 , 37 , 323 – 325 . DOI: 10.1002/(SICI)1521-3773(19980216)37:3<323::AID-ANIE323>3.0.CO;2-T
- [20] Tetrahedron 2000 , 56 , 6493 – 6496. DOI: 1016/S0040-4020(00)00613-X
- [21] Chem. Eur. J. 2002 , 8 , 3103 – 3114. DOI: 10.1002/1521-3765(20020715)8:14<3103::AID-CHEM3103>3.0.CO;2-C
- [22] Inorg. Chim. Acta 2002 , 337 , 287. DOI: 10.1016/S0020-1693(02)00998-2
- [23] Helv. Chim. Acta 1998 , 81 , 1223 – 1232. DOI: 10.1002/hlca.19980810533
- [24] J. Am. Chem. Soc. 2001 , 123 , 12907 – 12908 .124. DOI: 10.1021/ja012104v
- [25] Org. Lett. 2003 , 5 , 1563 – 1565. DOI: 10.1021/ol0343515
- [26] Org. Lett. 1999 , 1 , 837 – 839. DOI: 10.1021/ol990679f
- [27] Org. Lett. 2005 , 7 , 2881 – 2884. DOI: 10.1021/ol050834s
- [28] Org. Lett. 2003 , 5 , 217 – 219. DOI: 10.1021/ol027291w
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!


















No comments yet.