
本文作者:杉杉
导读
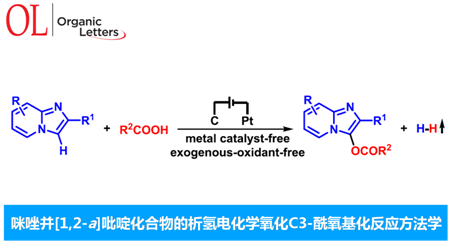
C3-官能团化的咪唑并[1,2-a]吡啶为有机化学中较为常见的含氮稠合杂环化合物,然而,对于咪唑并[1,2-a]吡啶的C3-酰氧基化方法学,目前尚未有文献报道。近日,武汉大学雷爱文教授课题组在Org. Lett.中发表论文,首次报道通过咪唑并[1,2-a]吡啶底物参与的电化学氧化C3-酰氧基化反应 (electrochemical oxidative C3 acyloxylation)方法学。值得注意的是,采用控制电流的策略,能够使上述的合成转化过程在更加温和的反应条件下进行。此外,上述的电化学氧化C3-酰氧基化策略能够成功应用于一系列芳香羧酸以及烷基羧酸底物。
Electrochemical Oxidative C3 Acyloxylation of Imidazo[1,2-a]pyridines with Hydrogen Evolution
Y. Yuan, Z. Zhou, L. Zhang, L. Li, A. Lei, Org. Lett. ASAP. doi: 10.1021/acs.orglett.1c02032.
正文
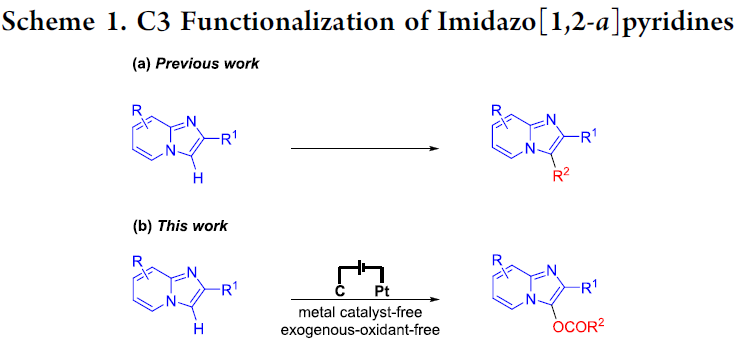
咪唑并[1,2-a]吡啶类化合物,尤其C3-官能团化的咪唑并[1,2-a]吡啶,为有机化学中较为常见的含氮稠合杂环化合物。并且,作为重要的结构单元,广泛存在于一系列天然产物、生物活性分子以及药物分子中,例如miroprofen、GSK812397、olprinone、zolimidine、minodronic acid以及alpidem。因此,合成化学家一直致力于咪唑并[1,2-a]吡啶C3-官能团化策略的研究。并且,在过去的几十年中,已经成功开发出多种能够实现咪唑并[1,2-a]吡啶C3-官能团化的反应策略,主要涉及芳基化[1]、烷基化[2]、羰基化[3]、卤化[4]、胺化[5]、膦酰化 (phosphonation)[6]、硒化 (selenation)[7]以及磺酰化 (sulfonylation)[8] (Scheme 1a)。然而,咪唑并[1,2-a]吡啶的C3-酰氧基化反应方法学的研究,至今尚未有文献报道。
近年来,有机电合成 (organic electrosynthesis)作为一种环境友好并高效的合成策略。通过阳极氧化或阴极还原过程,能够在无外源性氧化剂与还原剂存在的条件下,实现一系列有机分子的氧化还原过程。此外,通过电流或电压的调节,有机电合成策略能够实现在传统的化学氧化剂或还原剂条件下难以进行的合成转化过程。这里,基于作者前期对于析氢的电化学氧化交叉偶联策略的相关研究报道[9],武汉大学的雷爱文教授课题组首次报道采用咪唑并[1,2-a]吡啶参与的C3-酰氧基化反应方法学 (Scheme 1b)。值得注意的是,通过电化学阳极氧化策略,能够在无需加入金属催化剂或外部氧化剂的条件下,有效地实现各类咪唑并[1,2-a]吡啶底物的C3-酰氧基化过程。此外,作者通过析氢电化学氧化C-H/O-H交叉偶联策略的设计,使上述的C-H酰氧基化过程表现出优良的原子经济性与副产物的环境无害性。
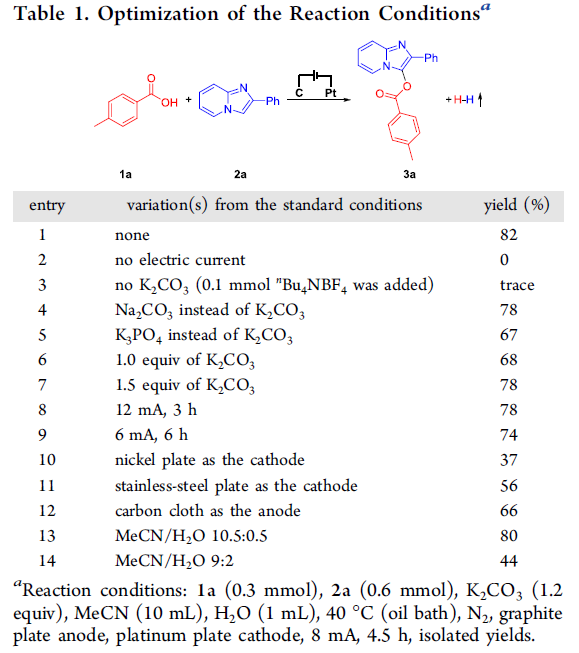
首先,作者采用对甲基苯甲酸1a与2-苯基咪唑并吡啶2a作为模型底物,进行相关反应条件的优化筛选 (Table 1)。确定最佳的反应条件为:采用K2CO3作为电解质与碱,石墨板 (graphite plate)作为阳极,铂板 (platinum plate)作为阴极,在乙腈与水的混合溶剂中,控制电流为8 mA的条件下,进行电解反应,最终获得82%收率的产物3a。
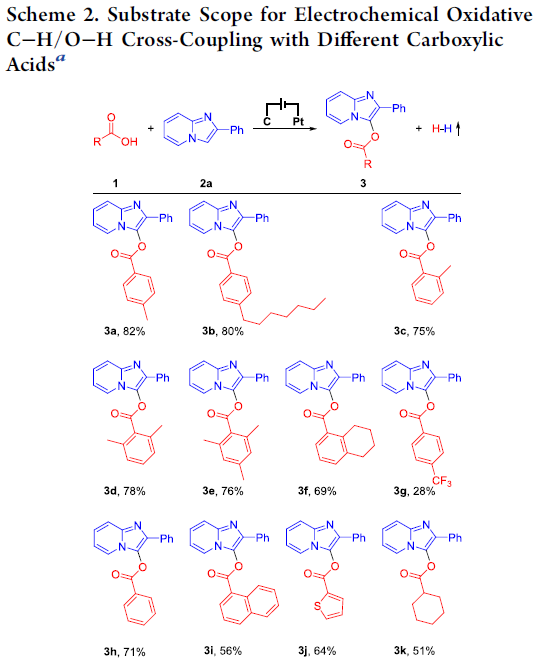
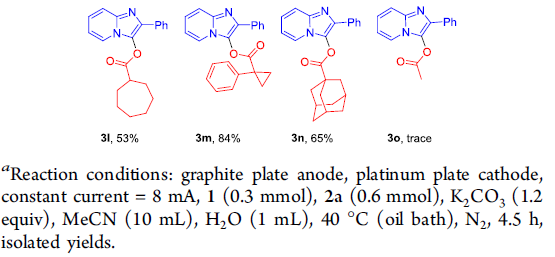
在上述的最佳反应条件下,作者首先对羧酸的底物应用范围进行考察 (Scheme 2)。研究表明,各类供电子基取代的芳香羧酸底物,均能较好地与上述的标准反应条件进行兼容,并获得相应的交叉偶联产物3a–3f,收率为69-82%。然而,对于具有吸电子基团取代的芳香羧酸底物,则仅能够获得较低收率的偶联产物,例如3g。之后,作者发现,采用苯甲酸,同样能够获得71%收率的偶联产物3h。同时,研究发现,1-萘甲酸与噻吩-2-羧酸同样能够有效地参与上述的偶联过程,并分别以56%与64%的收率,获得相应产物3i与3j。此外,作者进一步观察到,一系列烷基羧酸底物同样能够顺利地与2-苯基咪唑并吡啶(2a)进行反应,并获得相应的目标产物3k–3n,收率51-65%。
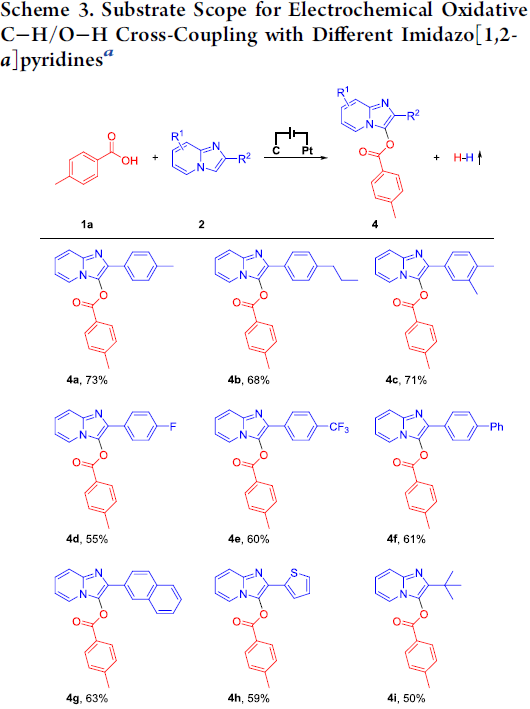
接下来,作者对咪唑并[1,2-a]吡啶的底物应用范围进行深入研究 (Scheme 3)。实验表明, R2为一系列具有供电子与吸电子基团取代的芳基时,均能够与上述最佳的反应条件良好地兼容,并获得相应的目标产物4a–4e,收率为55-73%。同时,R2为联苯基、萘基、噻吩基、叔丁基时,同样能够顺利地完成上述的电化学偶联反应过程,并获得相应的产物4f–4i,收率为50-63%。然而,上述标准反应条件对于C-2未取代的咪唑并[1,2-a]吡啶底物,则无法有效的实现上述的偶联反应过程,例如4j,这可能源自于相应自由基正离子中间体的不稳定性。此外,作者进一步观察到,采用6-Me-、6-OMe-以及8-F-取代的咪唑并[1,2-a]吡啶时,能够以中等至较高的反应收率,获得相应目标产物4k–4m。同时,该小组进一步发现,采用6-苯基咪唑并[2,1-b]噻唑底物时,同样能够获得相应的交叉偶联产物4n。

为阐明合理的反应机理,作者进行相关的CV(cyclic voltammetry)与控制实验研究 (Scheme 4)。首先,作者通过CV实验观察到,底物2a的氧化峰位于1.58 V,同时,1a 与K2CO3 混合物的氧化峰位于1.88 V。然而,在 0-2.5 V位置,未观察到显著的氧化峰。上述事实表明,与羧酸及其共轭碱相比,咪唑并[1,2-a]吡啶更易于氧化。之后,作者发现,2-苯基咪唑并吡啶 (2a)与苯甲酸在上述的标准条件下进行电解反应时,能够获得71%收率的目标产物3g,同时,能够获得14%收率的同偶联产物5a。进而表明,咪唑并[1,2-a]吡啶在控制电流电化学反应条件下,能够转化为相应的自由基正离子中间体。此外,作者发现,在无K2CO3存在的条件下,选择苯甲酸钠作为酰氧基化试剂与2-苯基咪唑并吡啶 (2a)进行上述反应时,能够获得41%收率的C3-酰氧基化产物3g (Scheme 4b),进而表明,K2CO3作用为去除羧酸中的质子,相关相应的羧酸负离子。

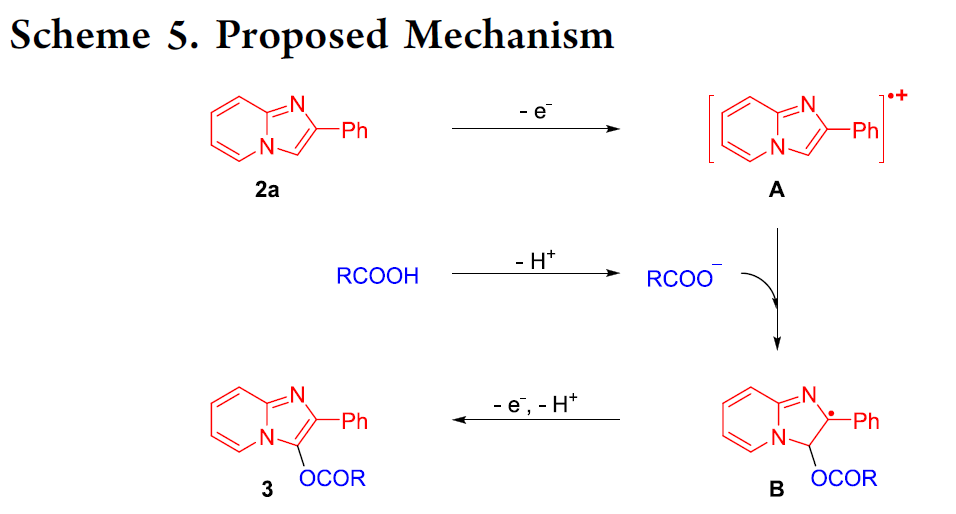
基于上述研究以及前期的文献报道[9c],作者提出一种合理的反应机理 (Scheme 5)。首先,通过2-苯基咪唑并吡啶 (2a)的阳极氧化过程,形成相应的自由基正离子中间体A。之后,在碱存在下,通过羧酸的去质子化步骤,形成RCOO−。同时,RCOO−进攻自由基正离子中间体A,进而形成自由基中间体B。接下来,在经历阳极氧化与去质子化步骤之后,最终获得相应的C3-酰氧基化产物3。同时,在阴极中,H2O经历相应的还原过程,形成OH–与H2。
总结
武汉大学雷爱文教授课题组首次报道一系列咪唑并[1,2-a]吡啶底物的电化学氧化C3-酰氧基化反应方法学。值得注意的是,通过控制电流的电化学策略,能够使上述反应在更加温和的条件下进行。同时,芳香族羧酸与烷基羧酸底物均能够与上述的标准反应条件良好地兼容。
参考文献
[1] A. K. Bagdi, M. Rahman, S. Santra, A. Majee, A. Hajra, Adv. Synth. Catal. 2013, 355, 1741. doi: 10.1002/adsc.201300298. [2] S. Jin, B. Xie, S. Lin, C. Min, R. Deng, Z. Yan, Org. Lett. 2019, 21, 3436. doi: 10.1021/acs.orglett.9b01212. [3] X. Zhao, E. Huang, Y. Zhu, J. Li, B. Song, X. Zhu, X. Hao, Org. Biomol.Chem. 2019, 17, 4869. doi: 10.1039/C9OB00596J. [4] D. R. Indukuri, G. R. Potuganti, M. Alla, Synlett 2019, 30, 1573. doi: 10.1055/s-0037-1611856. [5] S. Mondal, S. Samanta, S. Jana, A. Hajra, J. Org. Chem. 2017, 82, 4504. doi: 10.1021/acs.joc.7b00564. [6] F. Gao, K. Sun, X. Chen, T. Shi, X. Li, L. Qu, Y. Zhao, B. Yu, J. Org. Chem. 2020, 85, 14744. doi: 10.1021/acs.joc.0c02107. [7] Y. J. Kim, D. Y. Kim, Tetrahedron Lett. 2019, 60, 739. doi: 10.1016/j.tetlet.2019.02.001. [8] C. Breton-Patient, D. Naud-Martin, F. Mahuteau-Betzer, S. Piguel, Eur. J. Org.Chem. 2020, 2020, 6653. doi: 10.1002/ejoc.202001219. [9] (a) L. Lu, H. Li, Y. Zheng, F. Bu, A. Lei, CCS Chem. 2020, 2, 2669. doi: 10.31635/ccschem.020.202000512.(b) Q. Wang, X. Zhang, P. Wang, X. Gao, H. Zhang, A. Lei, Chin. J. Chem. 2021, 39, 143. doi: 10.1002/cjoc.202000407.
(c) Y. Yuan, A. Lei, Acc. Chem. Res. 2019, 52, 3309. doi: 10.1021/acs.accounts.9b00512.



















No comments yet.