

作者:石油醚
本期热点研究,我们邀请到了本文第一作者来自美国 Boston College的博士生徐士博为我们分享。
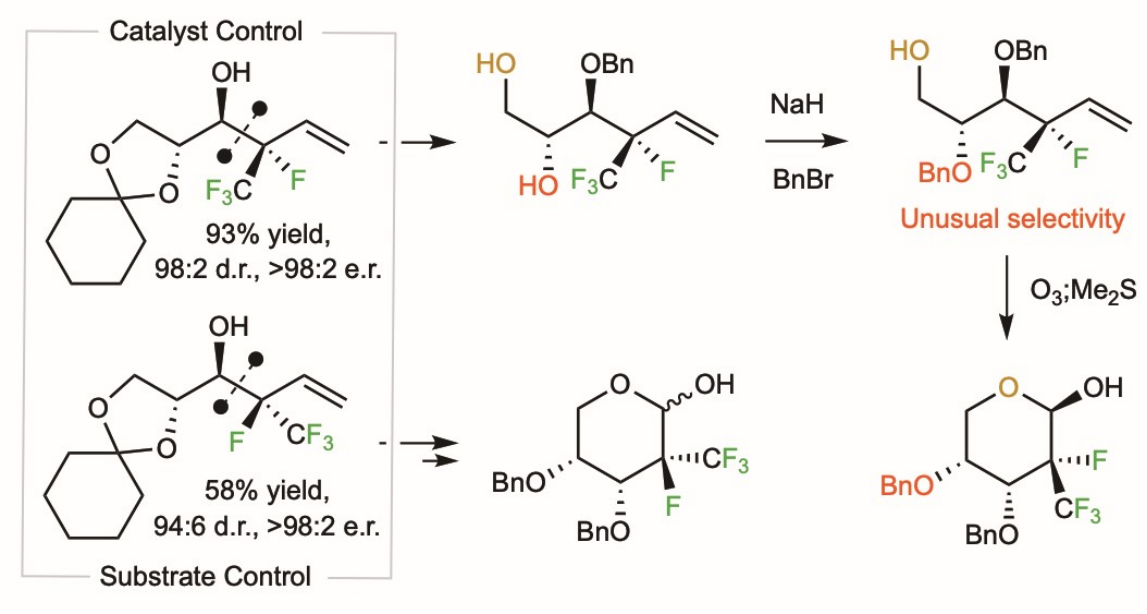
2022年11月14日,Nature Chemistry在线发表了来自美国 Boston College Prof. Amir H. Hoveyda团队题为「Diastereo- and enantioselective synthesis of compounds with a trifluoromethyl- and fluoro-substituted carbon centre」的研究论文。文章中,他们介绍一种多手性中心化合物高效引入氟原子和三氟甲基的新策略。我们展示了该方法在非天然核糖合成中的应用, 并利用催化剂控制和底物控制的互补策略实现立体发散合成。通过定点引入同时具备氟原子和三氟甲基的手性中心, 这个新方法潜在应用包括:(1) 拓展化学空间, 发现和制备新药物分子; (2) 改变有机分子构象并引入新反应性。
“Diastereo- and enantioselective synthesis of compounds with a trifluoromethyl- and fluoro-substituted carbon centre”
Shibo Xu, Juan del Pozo, Filippo Romiti, Yue Fu, Binh Khanh Mai, Ryan J. Morrison, KyungA Lee, Shaowei Hu, Ming Joo Koh, Jaehee Lee, Xinghan Li, Peng Liu* & Amir H. Hoveyda*
Nat. Chem.,2022, ASAP. doi: 10.1038/s41557-022-01054-4.
Q1. 请对“Diastereo- and enantioselective synthesis of compounds with a trifluoromethyl- and fluoro-substituted carbon centre”作一个简单介绍。
在我的导师 Hoveyda 教授的带领下, 课题组致力于开发新的催化剂和利用催化方法解决合成化学中的难题。氟原子和三氟甲基因为在药物合成领域的重要应用受到合成化学家的广泛关注。在抗癌抗病毒药物设计时, 核糖结构中引入氟原子尤其重要。高效, 并具有立体选择性引入氟原子的方法一直是手性催化的研究热点。此外, 作为自然界电负性最强的元素, 氟的吸电子能力和其高度电子局域的特性, 常常能改变有机分子的反应活性和选择性。
通过这次工作 [1], 我们介绍一种多手性中心化合物高效引入氟原子和三氟甲基的新策略。我们展示了该方法在非天然核糖合成中的应用, 并利用催化剂控制和底物控制的互补策略实现立体发散合成。通过定点引入同时具备氟原子和三氟甲基的手性中心, 这个新方法潜在应用包括: (1) 拓展化学空间, 发现和制备新药物分子; (2) 改变有机分子构象并引入新反应性。
Q2.在研究的时候遇到过怎样的困难呢?又是怎样克服的呢?
从2013年发现氨基酚有机小分子催化的不对称烯丙基化反应开始[2],课题组利用这个体系开发了诸多合成卤素取代的手性化合物的方法,并应用在了快速高效合成药物分子中。很快,我们的催化剂实现了商业化。在成功引入了三氟甲基,氯原子等缺电子基团后[3],下一挑战是活化同时具有氟原子和三氟甲基的亲核试剂,以合成具有这样一个独特手性中心的新化合物。尽管三氟甲基在药物设计中广泛存在,对手性中心同时具有氟和三氟甲基的化合物的研究非常稀少,原因是合成这类化合物的方法非常局限。我们决定攻克这个难题,一方面是在药物研发领域的潜在价值,另一方是科研工作者的好奇心:我们猜想四个氟原子在有限的空间内和临近的官能团相互作用,可能会给分子引入新的有用的性质。
这个课题是在我进组前就已经开始的。我们遇到的最大的困惑是反应机理。基于课题组对这个催化体系的认识和之前发表的工作[4],我们无法解释反应中观察到的区域选择性和立体选择性。为了进一步理解反应机理,我展开了包括动力学在内的一系列机理实验和反应副产物表征。在我和博后del Pozo博士的努力下,我们很快认识到这个反应的路径可能和其他氨基酚催化的烯丙基加成有所不同,由于氟和三氟甲基的同时存在,反应更有可能经过有机锌催化的路径,而不是之前提出的有机硼催化循环。
与此同时,我们开始了与匹兹堡大学(University of Pittsburgh)刘鹏教授课题组的合作,通过密度泛函计算探索反应机理的细节,并完善验证我们的猜想。经过近一年的合作努力,我们和刘教授的团队共同提出了有手性烯丙基锌参与的新催化循环,并解释了氟和三氟甲基的在控制区域选择性和立体选择性中关键作用。
得益于这个新方法学,我的同事Filippo Romiti博士和胡少伟博士分别合成了含2-氟-2-三氟甲基取代的核糖和六元环衍生物。为了展示方法的灵活性,我开始了这些分子的非对映体合成。在五元环非对映体的合成最后一步关环时我们遇到了选择性问题,不得不重新设计合成路线。在新一代合成路线中,我使用了一级醇选择新硅基化,并成功拿到了五元环产物。六元环非对映体合成的第一步最为困难:由于是底物控制过程,没有催化剂,再加上氟/三氟甲基取代的烯丙基硼试剂亲和性非常弱,醛的烯丙基化反应性很差。经过反应条件优化,我改良了Aggarwal课题组发表的烯丙基活化方法[5],解决了这个合成难题。
Q3. 本次研究主体,有没有什么让您感觉特别辛苦和烧脑呢?
对我来说,研究这个课题的过程非常有趣。
当然,也有比较辛苦的时候。在机理研究的阶段的早期,我们主要遇到了两个困难:一是基于组内从前的提出的反应机理,尽管我和同事们花了大量时间通过核磁实验,当量控制实验,络合物单晶表征的方法试图找到手性烯丙基硼中间体,但是没有没有找到任何实验证据;二是密度泛函计算表明,当氟和三氟甲基同时存在时,旧反应机理假设无法解释区域选择性和立体选择性。
针对这个困境,我和del Pozo博士提出的锌催化的可能新是一个非常大胆的假设,因为如果新假设成立,这个新的烯丙基化反应的反应路径就和课题组在2013年Nature[2]和2016年ACIE[4]中报道的机理完全不同。为了找到支持的证据,我和同事李星翰, del Pozo博士,一起设计了三组实验:一是动力学实验以确定锌,甲醇等物种在反应中可能的作用。为了拿到可靠的动力学数据,我连续一个月上夜班最大程度地利用有限的核磁资源。二是合成氨基酚-锌络合物单晶X射线衍射表征。我和同事们尝试了不同方法至少14次,最终拿到了一个二聚体结构,证明了锌盐可以和我们的配体形成稳定络合物。三是通过控制实验和核磁实验寻找催化剂反应中间体,理解催化剂失活的过程,产物抑制的机理,以及副产物鉴定。
在此期间,我们和刘鹏教授课题组通过在线会议和邮件频繁交流新发现。经过大量讨论和共同努力,同事付悦通过密度泛函计算优化了具体的手性烯丙基锌催化循环和立体模型,可喜的是计算结果和实验数据是吻合的。
非天然核糖全合成的过程时有惊喜和疑惑。我的同事胡少伟博士发现一个合成中间体二醇苄基化发生在位阻大的二级醇上。在胡博士离组工作后,我开始了对这个有趣的选择性的研究。我和导师Hoveyda教授讨论认为这个奇怪的选择性可能来源于分子中引入了氟和三氟甲基。验证这个猜想并不容易,我和同事Ryan Morrison博士经过一年多的探索合成了一系列完全不含氟或只含一个氟或三氟甲基的二醇衍生物。我们发现反应的选择性确实来源于氟和三氟甲基,无氟二醇苄基化优先发生在位阻小的羟基;只含一个氟或只含三氟甲基的二醇在苄基化反应条件下不稳定,容易消除或环化。多亏了我们和比兹堡大学刘教授课题组的深入合作,Binh Khanh Mai博士通过计算发现,氟和三氟甲基会显著提高周边氧负离子的亲核性,并高度极化临近烯基上的碳氢键使其具备氢键供体的性质。
你可能会问我为什么要关心一个非常特殊的二醇苄基化发生在哪个羟基?这个信息看起来很难延申到其它体系。我是这样认为的:二醇需选择性保护在合成中的意义只是一个方面。首先,得益于这个选择性,六元环核糖合成我们节省了两步保护脱保护,提高了效率。更重要的一个方面是我们通过研究选择性保护,揭示了氟和三氟甲基的协同作用可以给分子引入意想不到的性质。这个新发现可以延伸到改良生物活性分子的性质:如果在同一个碳上引入氟和三氟甲基,周边官能团的亲核性和成氢键的能力可能会发生显著的变化。
Q4. 将来想继续研究化学的哪个方向呢?
接下来有两个相关的方向值得进一步探索,一是拓展锌催化的认识到更有挑战性的体系,比如氟代烷基取代的氨基酸和氨基糖苷合成,二是研究其他氟代烷基基团对分子结构和分子性质的影响。作为合成化学科研工作者,我觉得每一个小发现都会加深我我们对化合物性质在分子层面的理解。我希望这个工作和以后的工作能够继续为药物设计,药物合成,尤其是抗病毒抗癌小分子领域提供一些新想法和新工具。
Q5. 最后,有什么想对各位读者说的吗?
对我来说, 科研的乐趣不仅在于好奇心和新发现,如果我的努力一点点工作更为人类更健康的未来做出轻微的贡献, 一切辛苦和付出都是值得的。
文献参考:
- [1] Xu, S. et al. Diastereo- and enantioselective synthesis of compounds with a
- trifluoromethyl- and fluoro-substituted carbon centre. Nat. Chem. ASAP. doi: 10.1038/s41557-022-01054-4.
- [2] Silverio, D. L. et al. Nature 494, 216–221 (2013).
- [3] Morrison, R. J. et al. J. Am. Chem. Soc. 142, 436–447 (2020).
- [4] Van der Mei, F. W. et al. Angew. Chem. Int. Ed. 55, 4701–4706 (2016).
- [5] Chen, J. L.-Y. et al. J. Am. Chem. Soc. 135, 5316–5319 (2013).
作者教育背景简介

教育背景:
2013-2017, 中国科学技术大学, 本科
2018- Boston College,博士
作者后记:
我来自内蒙古包头。2012年在全国高中化学竞赛中获得银牌,次年保送中国科学技术大学。在科大,我有幸加入恩师顾振华教授课题组,开始了探索有机合成和不对称催化的旅程。我对科研产的浓厚兴趣得益于顾老师的教导,他常常鼓励我探索发现新方法。在顾老师的栽培和师兄赵坤博士的指导下,我在本科阶段得以发表四篇SCI论文。能够有机会参与多个课题并熟悉不同的科研方向(天然产物全合成,铜催化高碘盐不对成开环,新型Catellani反应),我感到非常幸运。
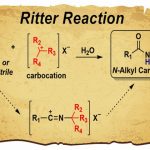
本科毕业后我来到美国波士顿学院Amir H. Hoveyda教授课题组攻读博士学位。Hoveyda教授既是一名杰出的科学家,也是关怀学生发展的导师。我从Hoveyda教授学到的不仅是是专业知识,更是重要的是作为科研工作者的思维方式,发现问题和解决问题的能力。博士最初的两年,在Juan del Pozo博士的指导帮助下,我的研究专注于铜催化不对称合成。我们利用廉价且稳定的腈类化合物实现了手性酮和三级醇的高效合成,成果发表在美国化学会志(JACS)。2020年我很荣幸被授予LaMattina Family Fellowship for Chemical Synthesis奖学金。目前我是五年级博士生,我的研究方向更着重于氟化学和天然产物骨架编辑的新策略。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载



















No comments yet.