
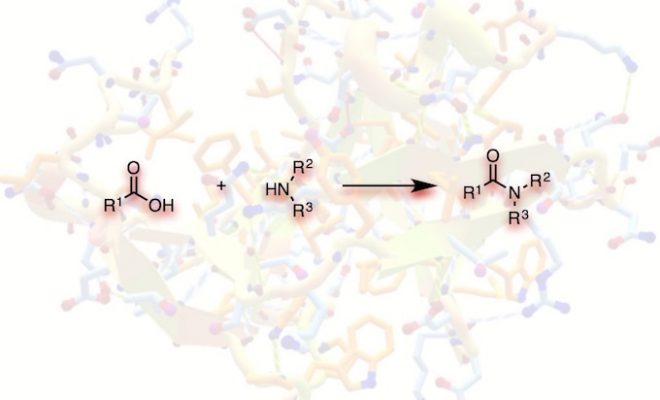
酰胺键是一类重要的官能团,不仅广泛存在于医药中间体,活性天然产物以及功能材料分子的结构之中,而且也是蛋白质结构中的重要化学纽带。酰胺键的构筑一直是方法学研究的活跃领域,传统的酰胺键的合成方法主要涉及到羧酸的活化,将其转化为酰卤,酸酐等衍生物后再与胺片段反应,或者利用各类缩合剂促进羧酸与胺的缩合,此外,近年来还陆续报道了诸如芳卤与胺在金属催化下构建酰胺键等新的策略。
酰胺合成研究背景及意义
酰胺的合成在药物化学研究中更是占有举足轻重的地位,近几年多篇对于药化领域的大数据分析论文都毫无例外地反映了这一点。比如从2016年AstraZeneca的Brown等[1]对2014年以及1984年两年中药化领域最常使用的合成反应进行的分析中可以发现,几乎同三十年前的情况相差无几,涉及酰胺合成的反应在结果中位列前茅。(见Figure 1.)
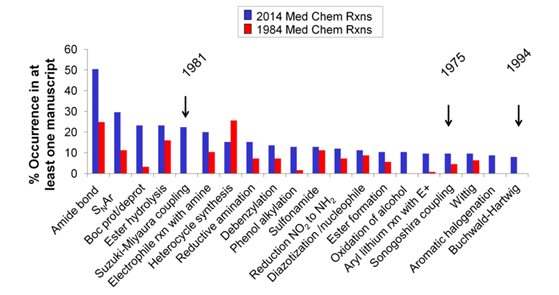
Figure 1.
2016年,Gilead和Pfizer的工艺研究人员共同在OPRD上发表了关于酸与胺反应生成酰胺的综述,[2]与之前其他已经发表的各类综述相比,该文的特色是所其评述反应的规模均达到100 mmol以上的反应量,可以说这篇综述是目前关于实验室或者工业中大量制备酰胺方法的最有价值的参考文献。文中列出位于统计前列的SOCl2,(COCl)2,EDCI, CDI等作为活化试剂的反应可以说是当前对于酰胺合成最为经典以及可靠的实验方法。(见Figure 2.)
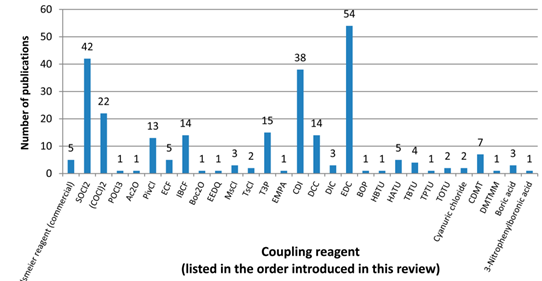
Figure 2.
酰胺合成面临的困难挑战
虽然化学家对于酰胺的合成方法已经有了相当数量的选择空间并且积累了足够的经验,但是仍然经常会在研究过程中出现目标酰胺的合成受阻或者不如预期效果的情况。很多时候这种困难在事先并不会被充分估计到,这也就是为何许多研究人员在文章中写到此处时都会如此描述——“This seemingly trivial transformation to produce XXX was plagued with poor isolated yields or low conversions”——至少说明在起初设计时,酰胺的合成在大部分研究者看来是完全不会有问题的。根据已有文献的报道以及小编本人在科研中的体会,发现在酰胺(或多肽)的制备中,除了对于手性底物缩合时容易发生消旋化这个问题之外,出现意外频率最高的是有Cα位存在大位阻羧酸以及缺电子芳胺参与的酰胺合成反应。下面,小编就将从近年来的几篇文献入手,结合其中的研究实例,来谈谈如何解决这个问题,也希望文献中提供的思路能对大家自己的课题有所借鉴。
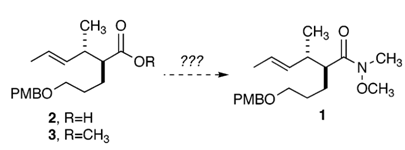
Scheme 1.
研究实例
2004年,Dake等[3]在对Nitiol的全合成研究中需要合成Weinreb酰胺1作为其中一个中间体以便下一步的研究,然而在合成中发现无论是以羧酸2还是甲酯3为前体参考已有文献进行转化(见Scheme 1.),原料或是不反应或是只得到低收率的产物以及混合物,反应结果不令人满意。作者推测应该是羧基周围庞大的空间位阻导致了该问题。通过调研文献,作者选择了MsCl和TEA为反应条件,通过与底物羧酸形成混合酸酐来达到活化的目的。在50 mg小试规模反应获得了80 %收率的良好结果,而直接以小试条件放大到克级规模时,由于PMB保护基脱除引起的副反应使收率降低为55 %。经过优化MsCl与碱的比例,该反应在1 g规模的收率顺利达到80 %。
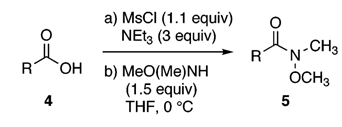
Scheme 2.
将优化后的反应条件(见Scheme 2)应用于其他立体障碍型羧酸底物4所对应的Weinreb酰胺5的合成,均获得了中等至良好的分离收率(见Table 1)。总的来说,此方法对于芳香或是脂肪类羧酸底物有良好的适应性,同时,足够温和的反应条件使得手性底物并未发生差向异构化。不过,需要指出的是,该方法合成Weinreb酰胺过程中容易形成N-甲氧基-N-甲基甲磺酰胺这个在TLC上不易检测的副产物,较难通过柱层析除去,作者尝试最成功的方法是利用加热真空干燥过夜,可以除去绝大部分该杂质。
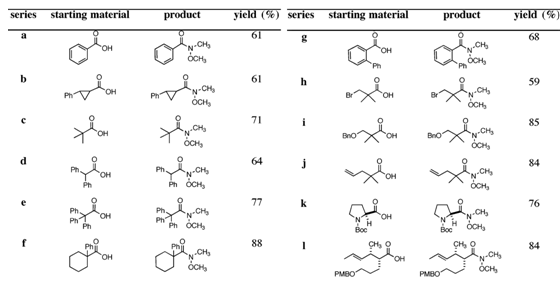
Table 1.
实际上,Dake等人所参考利用的活化方法,正是之前K.C. Nicolaou以及 Phil. S. Baran在CP分子全合成中所发展的方法,区别只在于, Baran以此活化了大位阻羧酸后制备的是α-重氮酮类化合物。[4]在CP分子的全合成中,Baran希望得到α-重氮酮7来进行Arndt-Eistert反应从而构建起目标分子中的季碳中心,但在从羧酸6制备7的过程中遇到了困难(见Scheme 3.)。考察6的空间结构可以发现,其羧酸基团周围的空间位阻障碍极其大,而羧酸又处于分子中凹的位置,导致常规的活化方式对其不起作用。例如以酰氯制成混合酸酐方法中,重氮甲烷对两个羰基位置都有进攻,而用Vilsmeir试剂在剧烈条件下尝试活化则导致了原料的分解以及甲酯的生成。在这种困境下,Baran必须要发展新的改良方法才能解决这个问题。Nicolaou和Baran意识到一个相对小的试剂,例如由MsCl和合适的碱原位生成的硫酰烯化合物(sulfene)也许能够穿透CP分子系统中的立体障碍而将羧酸转化为与酰氯具有类似反应活性的酰基甲磺酸酯(acyl mesylates)。事实证明,该方法确实有效,化合物6在用MsCl和TEA处理过后直接加入重氮甲烷反应,以高收率得到了目标产物7。
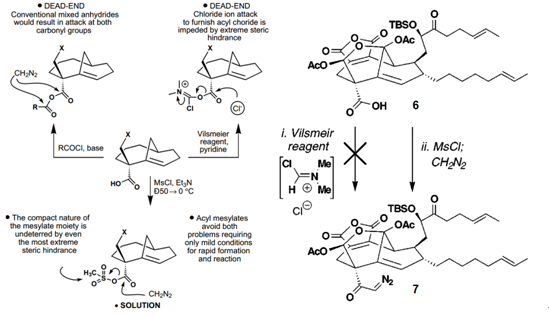
Scheme 3.
该方法操作简便,可以方便的制备大位阻的α-重氮酮类化合物。在CP分子的全合成过程中,Baran有机会将此方法在一系列类似的底物上进行尝试。CP全合成中的其他底物,带有硅烷保护基以及缩醛结构的对酸敏感的底物9–12,以及同样对碱敏感的底物8–10和13在该反应条件下均具有良好的耐受性,收率良好。在其他复杂的大位阻底物上利用该方法进行反应,效果同样令人满意,尤其是万古霉素衍生物19,在该分子中至少存在九个酰胺结构以及一系列敏感而易差向异构化的中心,底物依然顺利转化。(见Table 2.)
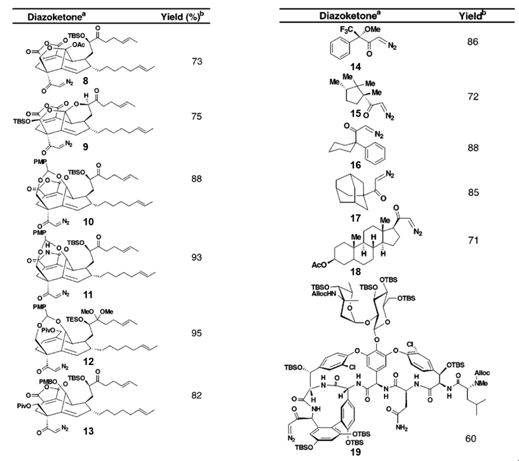
Table 2.
虽然之前文献中也有利用酰基甲磺酸酯的报道,但是文献中都没有给出确定的证据证明酰基甲磺酸酯就是实际的反应中间体。Nicolaou和Baran重现了一些声称利用酰基甲磺酸酯的文献内容,发现大部分反应中,磺酰氯介导的脱水生成的对称性酸酐才是真正的中间体。而Baran通过对本方法的中间体分离后并进行核磁共振表征,首次用光谱手段明确了酰基甲磺酸酯中间体的存在。
类似的,2011年上海医工院周伟澄等[5]在对拟肽类抑制剂进行研究时,借鉴此方法顺利完成了目标分子的合成。周等最初的尝试是以Cbz保护的Leu,Ile和Phe三种氨基酸为合成砌块分别与芳胺进行缩合,然而效果不佳,常见缩合剂手段给出的收率较低,无法满足进一步的研究需要。经过推测,周认为是N-保护氨基酸的位阻以及芳胺的低亲核性同时导致了低收率。在尝试MsCl与碱配合活化羧基方法时,以三乙胺或是吡啶作为碱给出的结果同样欠佳。通过对碱进行筛选,周发现利用N-甲基咪唑可以改善反应结果,(见Table 3.)在对反应条件进行优化之后,三种大位阻的氨基酸与缺电子的芳胺的缩合收率大幅度提高。
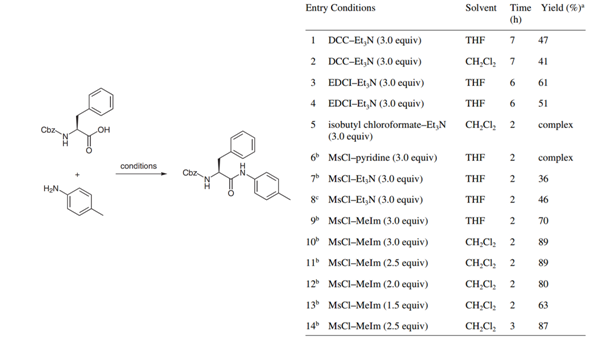
Table 3.
2004年,Goldman在前期将羧酸金属盐类比有机酯并作为酰化剂与胺的反应基础上,从Zr(IV), Ti(IV)和Ta(V)中选出最优的Ta(V)进一步研究,发现TaCl5可以介导大位阻羧酸与胺之间的酰胺键形成,并且在手性羧酸底物参与的反应中只有很低水平的消旋化发生,可能的反应机制如Scheme 4所示。[6]

Scheme 4.
与之前大多的脱水性缩合思想不同,2012年Bode提出了利用格氏试剂与异氰酸酯反应构建酰胺键的新思路。[7]该方法反应条件简便,对于格氏试剂片段或者异氰酸酯片段均有广泛的底物适应性与兼容性,可以高效地制备大位阻与缺电子酰胺。(见Table 4.)当底物中存在酯基,羰基等可能与格氏试剂反应的潜在竞争性的官能团时,所得目标产物的产率依然很高。值得注意的是,使用该方法时需要考虑所用原料转化为格氏试剂与异氰酸酯的可行性问题。
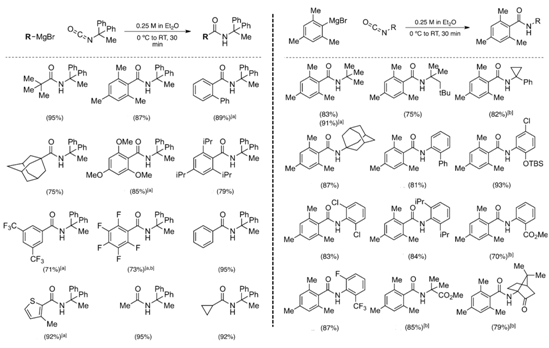
Table 4.
2016年,Ulven等在重复文献过程中,[8]根据文献条件以HATU为缩合剂,将20与21缩合制备22时反应进行不佳,进一步尝试其他缩合方法也无功而返。(见Table 5.)该反应的难点在于芳胺的低亲核性及位阻与羧酸周围的位阻且存在敏感的叔丁酯基团。
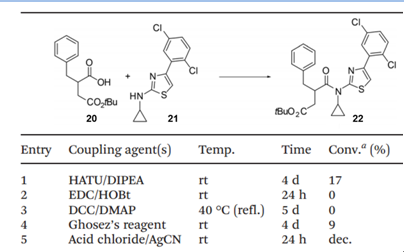
Table 5.
Ulven考虑到需要将缩合过程中的位阻因素降低到最小才可能使反应成功进行,于是决定引入体积较小的酰氟。经过比较之后,决定使用没有毒副产物的BTFFH作为酰氟化试剂。利用该方法不仅成功合成了原来的目标产物,而且在原料的类似物以及已有文献报道的低收率大位阻底物案例中同样取得了良好的结果。(见Table 6.)不过,该方法在缺乏第二个α取代基的芳基乙酸类底物以及更大位阻的某些底物上效果不佳,而当大量制备时,BTFFH的成本也是需要考虑的问题。
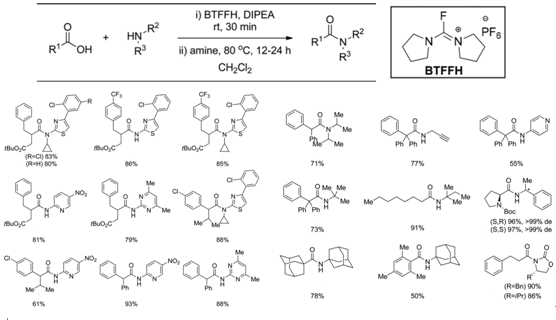
Table 6.
从以上几篇分享的文献中可以看出,对于大位阻的底物,尝试寻找较小体积的活化策略往往是比较好的办法,并且反应的条件需要考虑底物的耐受性最好尽量温和。而当已有方法不能取得较好结果时,通过梳理文献进行适当的改进也能取得不错的效果。希望以上几点总结能给各位读者在日常科研生活中提供一点帮助。
参考文献
- Brown D G, Boström J. [J]. J. Med. Chem., 2016, 59, 4443~4458.
- Dunetz J R, Magano J, Weisenburger G A. [J]. Org. Process Res. Dev., 2016, 20, 140~177.
- Woo J C S, Fenster E, Dake G R. [J]. J. Org. Chem., 2004, 69, 8984~8986.
- Nicolaou K C, Baran P S, Zhong Y, et al. [J]. Org. Lett., 1999, 1, 883~886.
- Mao L, Wang Z, Li Y, et al. [J]. Synlett., 2011, 129~133.
- Fang J B, Sanghi R, Kohn J, et al. [J]. Inorg. Chim. Acta., 2004, 357, 2415~2426.
- Schäfer G, Matthey C, Bode J W.[J]. Angew. Chem. Int. Ed., 2012, 51, 9173~9175.
- Due-Hansen M E, Pandey S K, Christiansen E, et al. [J]. Org. Biomol. Chem., 2016, 14, 430~433.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!










No comments yet.