
作者:石油醚
导读
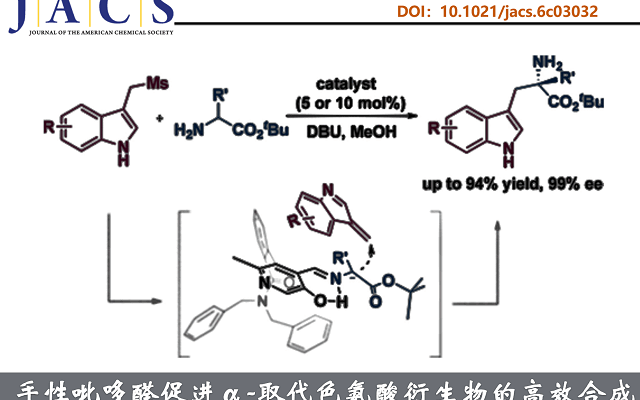
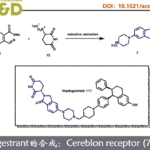
近日,上海师范大学赵宝国教授和肖晓教授团队在J. Am. Chem. Soc.上发表了题为[ Chiral Pyridoxals Enabled Efficient Enantioselective Synthesis of α-Substituted Tryptophan Derivatives ]论文。赵宝国教授和肖晓教授团队以联苯基轴手性吡哆醛作为催化剂,成功实现了吲哚烯亚胺与N-未保护氨基酯发生不对称加成,开发了一种简洁、模块化的合成策略,高效制备α-四取代色氨酸酯。
“Chiral Pyridoxals Enabled Efficient Enantioselective Synthesis of α-Substituted Tryptophan Derivatives
Dongchen Cai, Yunfei Huang, Tongyin Chen, Siqi Liu, Xiao Xiao* and Baoguo Zhao*
J. Am. Chem. Soc.2026, ASAP. DOI: 10.1021/jacs.6c03032”
正文:
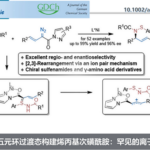
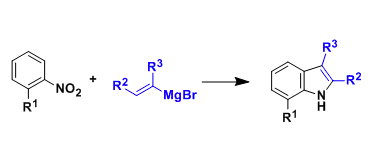
色氨酸及其衍生物在化学、生物学与医药研究中具有不可替代的战略地位(Scheme 1A)。目前,吲哚环(N1及C2–C7位)的官能化已取得显著进展,包括适用于肽等复杂分子的 late-stage 修饰策略。然而,氨基酸骨架α-位的结构修饰仍然严重滞后(Scheme 1B,模块II)。但现有α-官能化方法极少,仅限于少数不对称催化体系,如手性磷酸或硫脲催化氮杂内酯对3-乙烯基吲哚/芳基磺酰吲哚的加成,以及金属/手性醛协同或双金属催化的不对称Heck–烷基化反应。此外,α-取代色氨酸可显著限制构象,从而调控生物活性、代谢稳定性与药代动力学性质。目前,已有多个α-取代色氨酸类药物获批或进入临床,如CCK-B拮抗剂CI-988、GRP-R拮抗剂PD-176252和NK1拮抗剂PD-174424(Scheme 1A)。因此,亟需发展通用、高效、高对映选择性的新策略,直接从简单原料构建α-取代色氨酸衍生物。
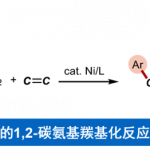
α-四元色氨酸类似物由两部分构成:含吲哚的骨架(模块I)和N-未保护的α-取代甘氨酸支架(模块II)。二者模块组装构建新的C-C键,是合成此类化合物最高效、最简洁的策略。其中,吲哚烯亚胺是模块I中理想的亲电模板。而模块II中的N-未保护α-取代甘氨酸衍生物则面临根本性挑战:α-碳空间位阻大、亲核性弱,且游离NH₂基团易发生竞争反应,严重阻碍其碳负离子对亚胺烯受体的加成。羰基催化为解决上述挑战提供了一种根本策略。
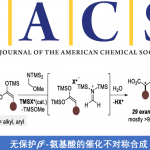
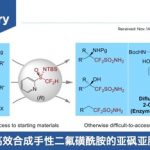
近些年来,上海师范大学赵宝国教授团队长期从事于羰基催化未保护伯胺的不对称α-C-H键官能团化研究,成功实现活化伯胺,如α-氨基酸酯,与不同的亲电试剂的多种不对称转化,例如甘氨酸酯的不对称仿生Mannich反应(Science. 2018, 360, 1438)、仿生Aldol反应(Angew. Chem. Int. Ed. 2021, 60, 20166)、Michael加成反应 (Angew. Chem. Int. Ed. 2021, 60, 10588)、不对称烯丙基化反应 (Angew. Chem. Int. Ed. 2022, 61, e202200850) 、丙氨酸酯的不对称烷基化反应 (ACS Catal. 2023, 13, 9150-9157)以及硝基烯烃的直接不对称α-C加成反应( Angew. Chem. Int. Ed. 2025, e202506342.),得到一系列手性氨基酸衍生物。对于惰性伯胺如苄胺和炔丙基胺,也成功实现了它们对高活性亲电试剂(如醛、三氟甲基酮和α,β-不饱和酮)的不对称加成 (Nat. Catal. 2022, 5, 1061-1068; Angew. Chem. Int. Ed. 2022, e202206111; J. Am. Chem. Soc. 2024, 146, 38, 25927–25933;Nat Catal. 2025 8, 668–677))。此外,利用光/钯/吡哆醛三重协同催化策略,成功实现氨基酸酯不对称α-烯丙基化(Angew. Chem. Int. Ed. 2025, 64, e202418910)。
近日,赵宝国教授和肖晓教授团队以联苯基轴手性吡哆醛作为催化剂,成功实现了吲哚烯亚胺与N–未保护氨基酯发生不对称加成,开发了一种简洁、模块化的合成策略,高效制备α-四取代色氨酸酯。DFT支持的机理研究表明,立体选择性主要由空间位阻效应控制。相关工作发表于“J. Am. Chem. Soc.”上。
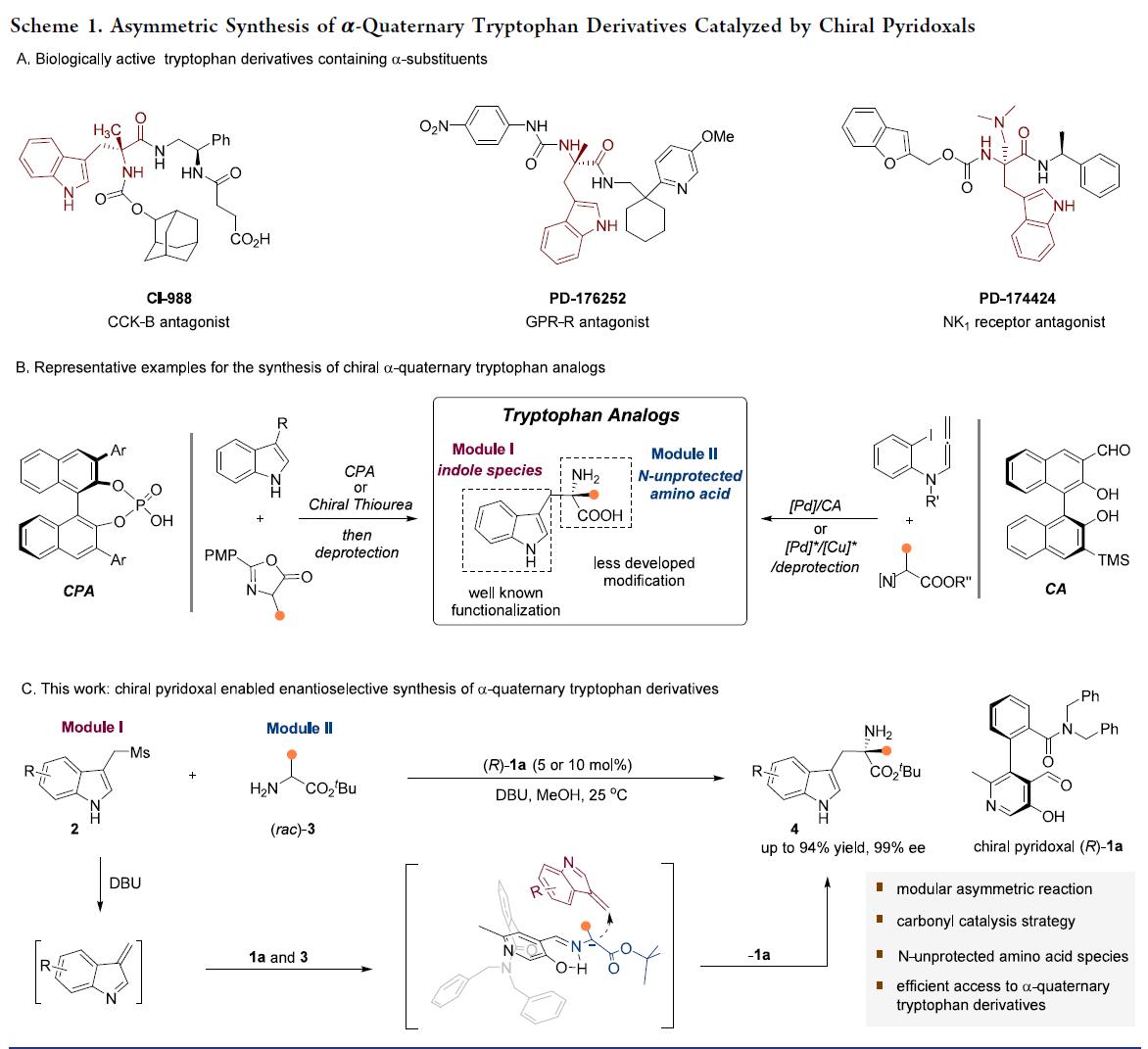
首先,作者以吲哚衍生物(2a)和α-甲基甘氨酸酯(3a)作为模型底物对反应条件进行了筛选(Scheme 2)。研究结果表明:1)碱性较强的有机碱(DBU、DBN、TMG、TBD)效果最优,以80-96%的产率和98%ee获得相应产物(entries 2–4),碱性较弱的有机碱(DABCO、Et₃N)难以驱动2a生成活性吲哚烯亚胺中间体对反应无效(entries 5–6),而Cs₂CO₃可反应,但产率中等(entry 7);2)醇类溶剂(MeOH、EtOH、iPrOH)的效果最佳(entries 8–11),因2a在 THF、CH₃CN或甲苯中的溶解性较差导致不反应(entries 12–14);3)对甲苯磺酰基(Ts)和苯磺酰基(PhSO₂)优于甲磺酰基(Ms)。因其更强的吸电子性加速烯亚胺形成,导致α-氨基碳负离子捕获滞后,引发甲醇加成等副反应,产率下降(entries 15–16);4)联苯基手性吡哆醛(1a)高效催化反应,以96%的产率和99% ee获得产物(entries 17–22),中心手性类似物(1b)、联萘基手性吡哆醛(1c)几乎不反应(entries 17–18)。增大侧链位阻(1d、1e)降低产率(entries 19–20)。N-甲基手性吡哆醛(1f)和氨基醇型催化剂(1g)活性低、对映选择性差(entries 21–22),进而表明,该转化无需更高亲电性的醛基,也不依赖氢键供体侧链。
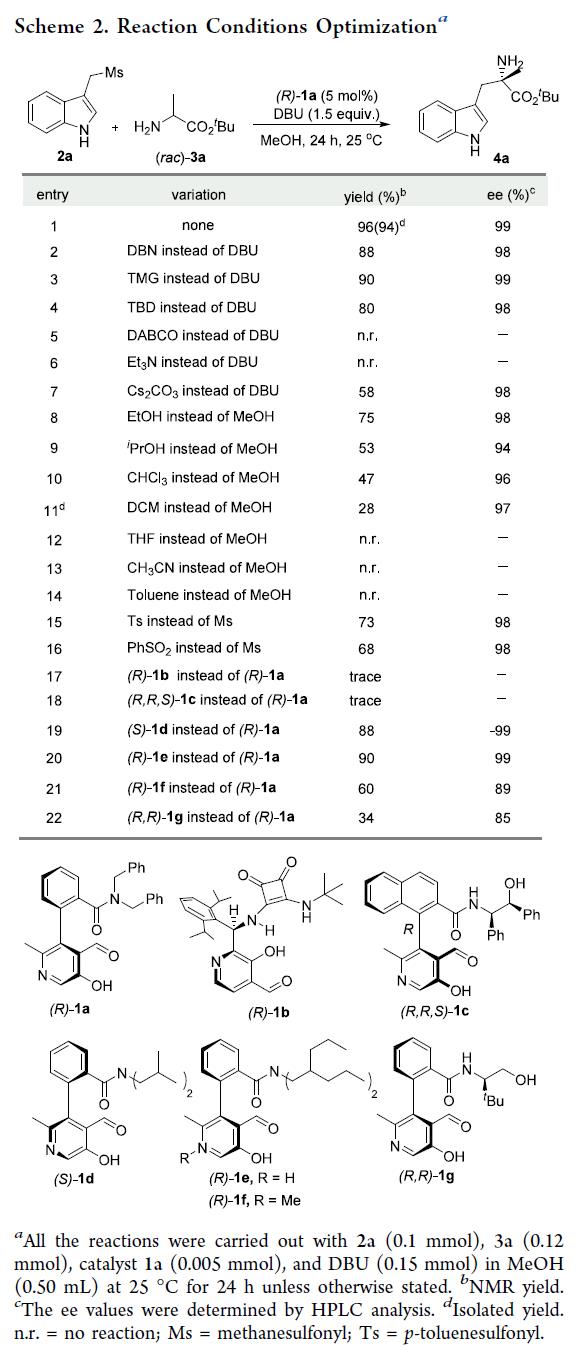
在最优条件下,作者对底物的兼容性进行了考察(Scheme 3)。一系列不同的吲哚衍生物均可与α-甲基甘氨酸酯(3a)反应,以40-94%的产率和95-99% ee获得相应α-取代色氨酸衍生物(4a-4l)。产物收率58%–92%,ee值95%–99%。除了α-甲基甘氨酸酯(3a)之外,其他α-烷基(4m–4q)、芳基(4r–4t)、杂原子(4u–4y)、环烷基(4z–4ab)、缩醛(4ac)、烯烃(4ad)、酯(4ae)取代的甘氨酸酯与2同样可以兼容(4m-4ai)。如烷基等官能团。此外,含天然产物或药物骨架的甘氨酸酯也可反应,如亚油醇(4af)、香茅醇(4ag)、诺波尔(4ah)、奥沙普秦(4ai)等。
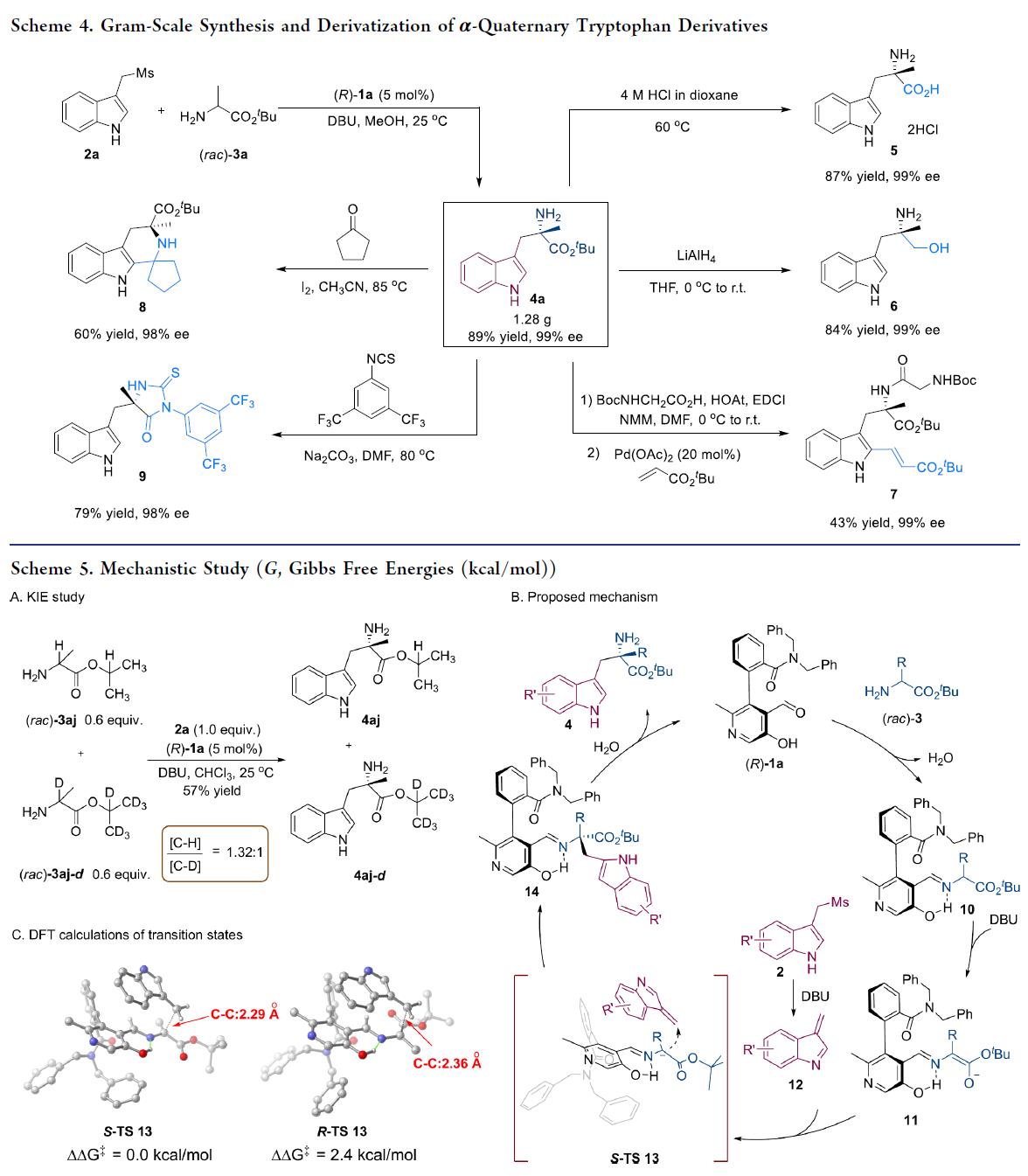
接下来,作者对该反应的实用性进行研究,如Scheme 4所示。即1)克级规模实验:89%产率和99% ee获得α-甲基色氨酸酯4a的克级合成;2)4a在碱性条件下水解获得手性α-甲基色氨酸5;3)LiAlH₄还原4a得手性氨基醇6;4)吲哚C2位C−H烯基化得化合物7;5)4a与环戊酮缩合得稠环化合物8;6)4a与芳基异硫氰酸酯反应得生物活性硫代咪唑烷酮9。
最后,作者一系列实验和DFT计算对反应机理进行了研究(Scheme 5)。初步实验表明:KIE实验以及初始反应速率测定实验表明:α-C−H键断裂未参与决速步(Scheme 5A)。机理研究表明(Scheme 5B):催化剂1a与氨基酯3缩合形成亚胺10,经DBU脱质子生成烯醇中间体11;后者对原位生成的吲哚烯亚胺12进行共轭加成,经过渡态S-TS13得到中间体14;水解后释放产物4并再生1a。DFT计算表明,S-TS13比R-TS13低2.4 kcal/mol(Scheme 5C),进而解释反应的对映选择性。立体控制源于吲哚烯亚胺从上方进攻11,避开(R)-1a酰胺侧链及甘氨酸叔丁基的空间排斥,从而专一构建(S)-构型。
总结
赵宝国教授和肖晓教授团队以手性吡哆醛作为催化剂,在温和条件下实现了吲哚烯亚胺与N-未保护α-取代甘氨酸酯之间的不对称加成,构建了一系列色氨酸衍生物。该策略建立模块化、高效的合成平台,彰显羰基催化构建复杂手性胺的独特能力。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/














No comments yet.