
本文作者:Azathoth 写作指导:Cyclization Xu
导读:
本文介绍了锰催化氢化领域的研究进展。文中描述了不同类型锰催化剂的设计与性能,通过分析不同配体和反应条件对催化效率和选择性的影响,展示了锰催化剂在不对称氢化反应中的潜力。清华大学刘强团队提出了关于锰催化氢化的多种活化模型,并将其应用在催化剂的设计上,推动了该领域的快速发展。
正文:
锰是仅次于铁、钛,储量排第三位的过渡金属,具有很大的应用价值。提到锰催化的不对称反应,我们首先想到的就是课本上的Jacobsen–Katsuki不对称环氧化。但是均相锰催化的氢化一直到2016年才开始发展,属于一个比较新的领域。目前的已知的锰催化氢化都是外球机理,对于外球机理的金属催化不对称氢化而言,反应最重要的两步是金属氢物种的生成和氢负的转移。但这两步的影响因素往往是相互矛盾的,金属氢物种的活性越高,氢负越容易转移,然而也意味着金属氢物种也就越难生成。对于钌铑等靠后的过渡金属而言,它们的金属氢物种生成和氢负转移恰好达到了一个平衡,金属氢物种既容易生成也容易与底物发生反应。而对于第一行过渡金属而言,金属氢物种的活性非常高,虽然这意味着强大的反应活性,但同时也导致了金属氢物种非常难以生成。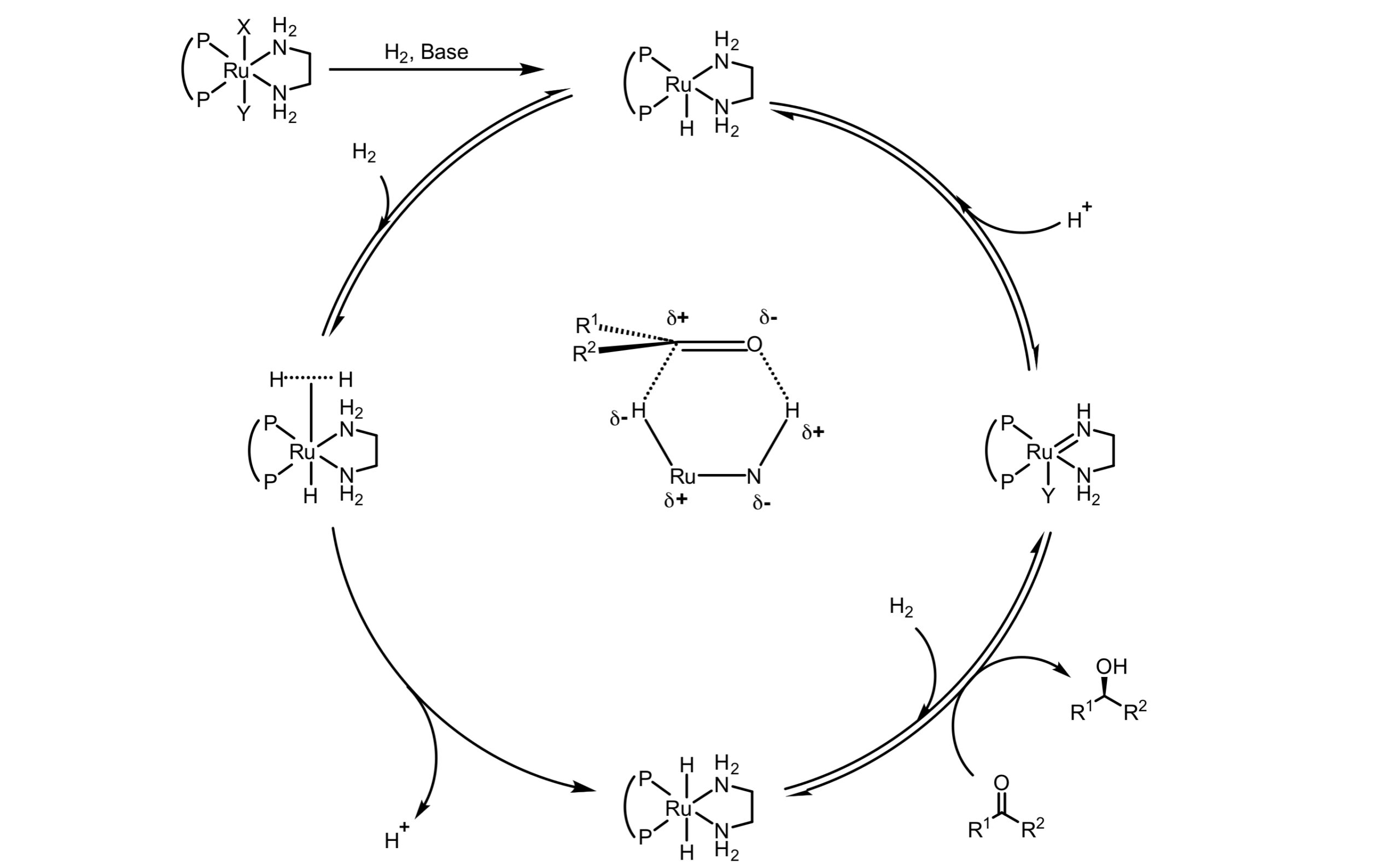
Fig.1. Typical outer-sphere mechanism catalytic hydrogenation
目前公认最早的均相锰催化氢化是由Beller 等人实现的[1],他们用PNP三齿配体和锰的络合物实现了苯腈、醛、酮的氢化。Beller的研究表明,锰催化剂必须要有一氧化碳配位,通过反位效应来稳定锰-氢键,否则锰氢物种的能量会非常高,难以生成,从而无法实现催化氢化。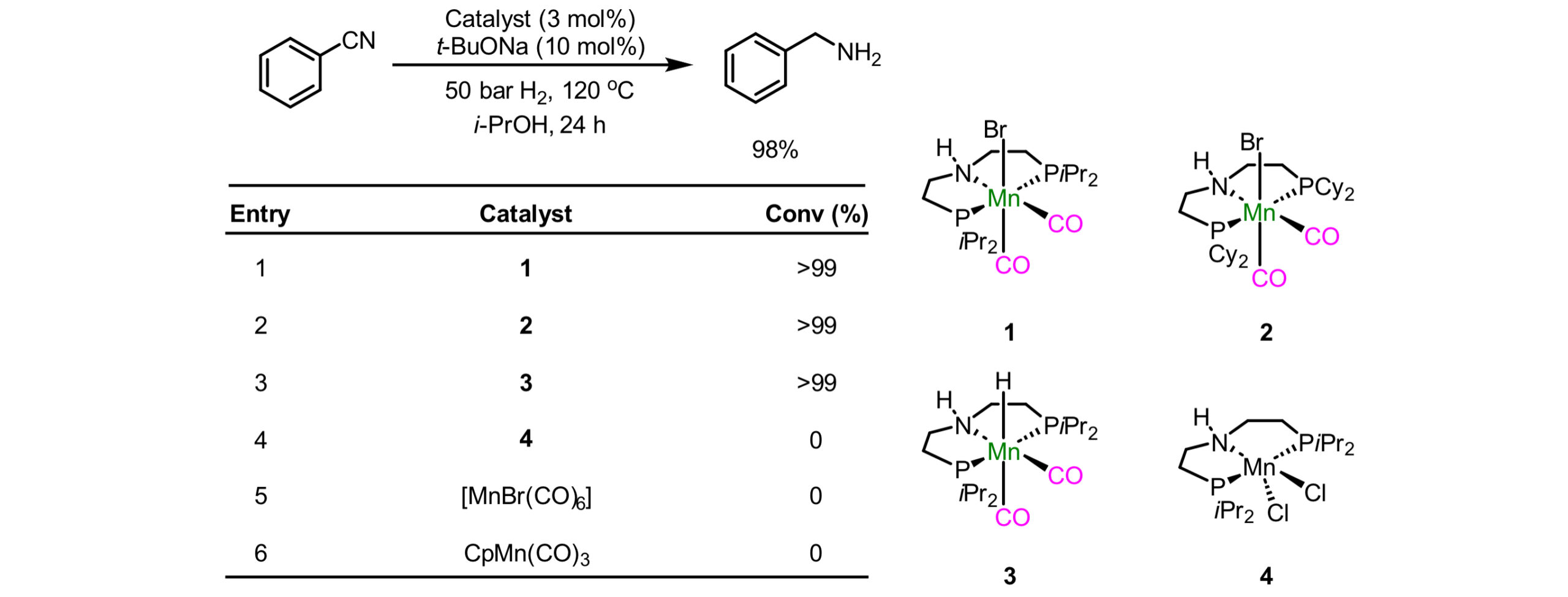
Scheme 1. One of the earliest homogeneous manganese-catalyzed hydrogenation reactions: hydrogenation of nitriles.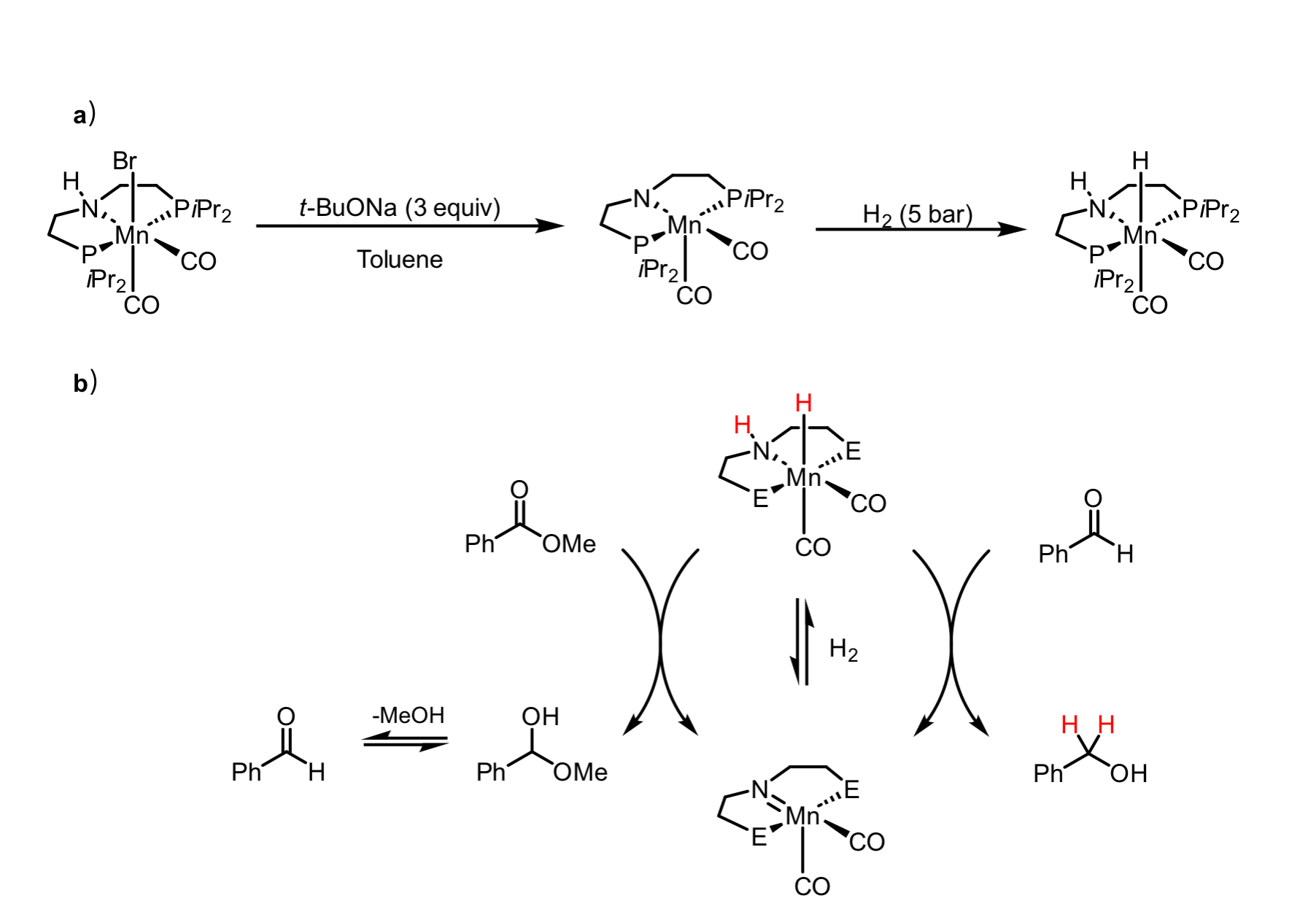 Fig.2. Mechanism of manganese-catalyzed hydrogenation
Fig.2. Mechanism of manganese-catalyzed hydrogenation
影响反应活性无外乎两个主要原因:空间位阻和电子效应。对于均相的锰催化氢化反应而言,在一定程度上更富电子、更小位阻的催化剂拥有相对更高的活性。小的位阻有利于催化剂与底物接近,而富电子有利于提高金属氢物种的亲核能力。如Kempt的工作所示,当吡啶配位基团的对位有氨基取代时,催化剂的活性会得到提升[2]。Beller后续的工作也显示了位阻对催化剂活性的影响[3]。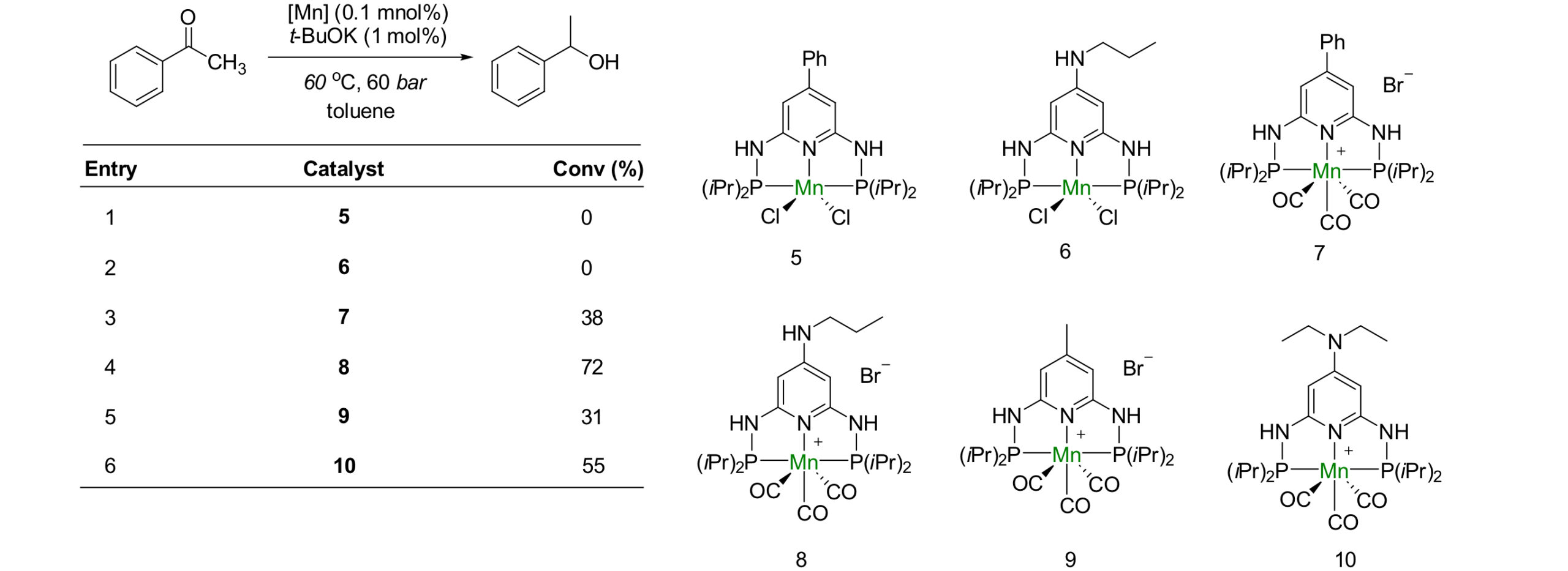 Scheme 2. The enhancement of the electron-donating ability of the ligand is beneficial for improving catalytic activity.
Scheme 2. The enhancement of the electron-donating ability of the ligand is beneficial for improving catalytic activity.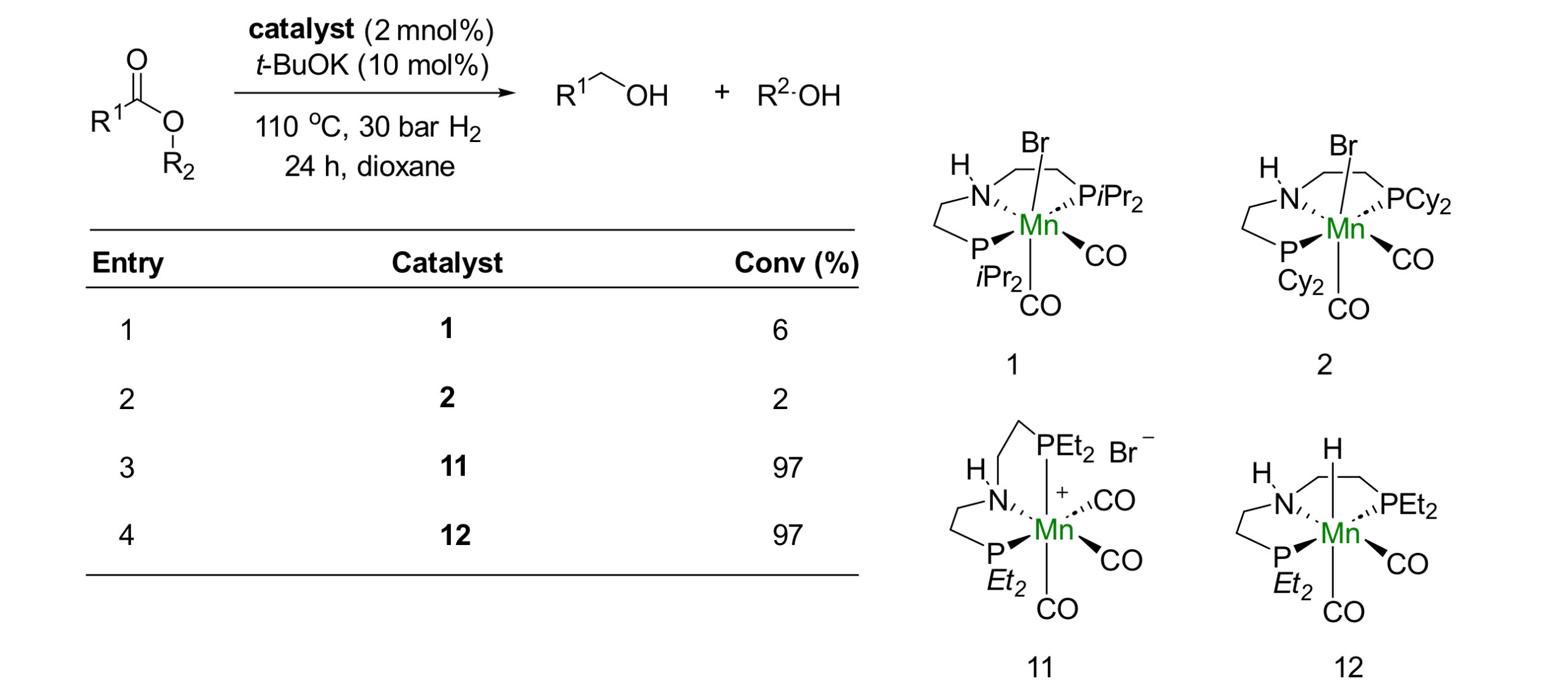
Scheme 3. The reduction of ligand steric hindrance is beneficial for improving catalytic activity.
Beller 、Clarke、Rueping、Balskus等人分别以咪唑、吡啶为配位基团设计了PNN配体[4] [5] [6],PNN-Mn的催化活性相对于PNP-Mn催化剂有巨大的提升。PNN-Mn催化剂可以氢化如酰胺、酯和碳酸酯这些具有很大挑战性的底物,这是很多PNP-Mn催化剂难以实现的。
 Scheme 4. In most cases, PNN ligands exhibit higher reaction activity than PNP ligands.
Scheme 4. In most cases, PNN ligands exhibit higher reaction activity than PNP ligands.
后来清华大学刘强和重庆大学蓝宇解释了PNN-Mn络合物活性强于PNP-Mn络合物的原因:他们认为PNN配体相对于PNP配体的优势在于位阻小、富电子,大大降低了氢化反应的能垒[7]。
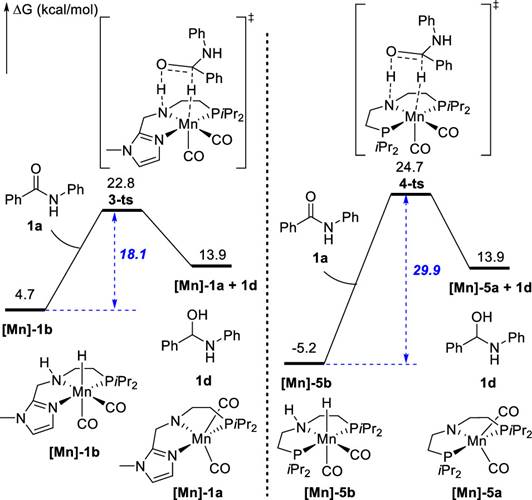 Fig.3. Reasons for higher activity of PNN ligands: reduced steric hindrance, electron-rich nature, lowering the energy of substrate-catalyst adducts, facilitating easier reactions.
Fig.3. Reasons for higher activity of PNN ligands: reduced steric hindrance, electron-rich nature, lowering the energy of substrate-catalyst adducts, facilitating easier reactions.
随着以上研究的深入,他们通过分离中间体进行氢化实验等方式发现,配体与碱形成氮负离子-碱金属盐之后往往反应活性更高。因为碱金属离子可以稳定金属氢物种与底物的加合物,而且负离子的形成还能使得配体更富电子,增强了金属上氢负的亲核能力,实现了碱金属离子介导的π轨道方向活化[8]。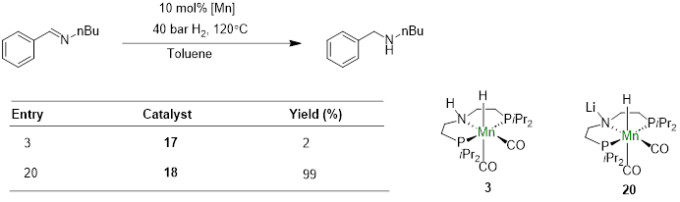
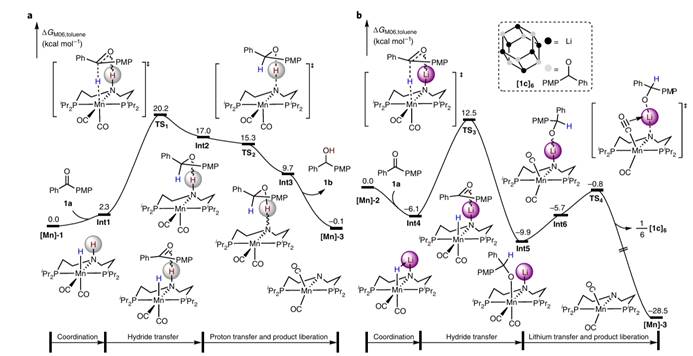 Fig.4. Alkali metal ion-mediated π-activation with the C=N/C=O double bond in the substrate,
Fig.4. Alkali metal ion-mediated π-activation with the C=N/C=O double bond in the substrate,
使用Lewis酸或质子酸来活化极性双键是非常常见的做法,在外球机理模型和碱金属离子介导的π轨道方向活化模型中,催化剂中的质子氢或者碱金属离子直接与底物极性双键中的氮/氧作用,有作为酸活化极性双键的可能性。但是上述两种模型中质子氢或碱金属离子通常只能和底物的π轨道方向而不是σ-轨道方向作用,难以发挥出酸的功能。基于此清华大学刘强提出了σ-诱导活化[9],即通过调控金属阳离子,使其与底物氮/氧的σ-键方向孤电子对作用,提高了底物的亲电能力,大大提升了氢负转移的效率。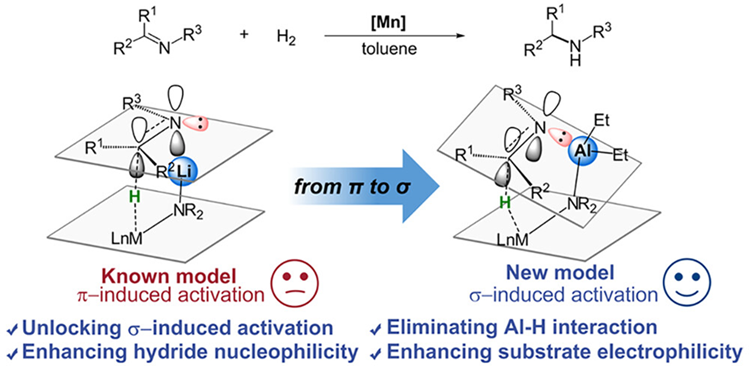
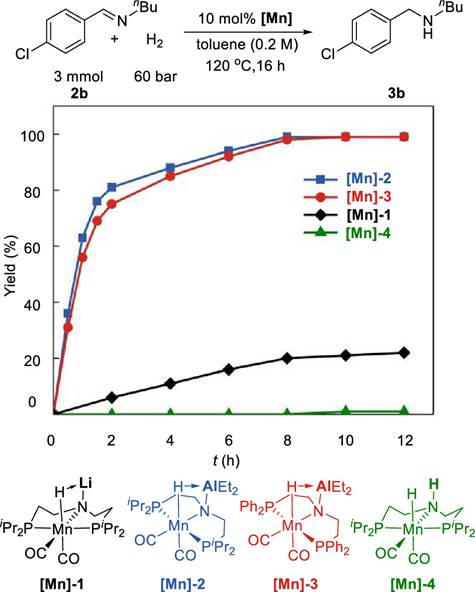
Fig. 5. Comparison of catalytic activity of HMn-NH, HMn-NLi, and HMn-NAl systems.
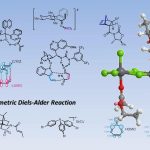
上述机理上的铺垫启发着科学家们对于不对称反应的探索。向锰络合物中引入手性元素,便有机会实现锰催化的不对称氢化。向锰络合物中引入手性元素主要有两种方式,Beller、丁奎岭等[10] [11]将配体膦的部分做成手性的BPE片段,得到了经式配位的锰络合物;Kirchner、刘强[12] [13] [14] [15]等则以二茂铁为基础,由二茂铁引导配体在金属上发生面式折叠,实现配体面手性到金属的中心手性再到产物手性的传递。
Fig. 6. Some design of chiral ligands
刘强课题组根据“小位阻,富电子”这一原则发展了高活性的PNN-Mn络合物,实现了喹啉、3,3-二取代3-H吲哚、喹喔啉等底物的不对称氢化[12] [13] [14]。
在对喹啉进行不对称氢化时,配体上的N-H部分可以通过金属-配体协同作用显著影响催化氢化反应的反应性,当配体烷基氮上的氢被甲基取代时,催化剂的反应活性急剧下降。而咪唑氮上的氢被甲基取代时反应活性和选择性也发生了下降。咪唑与喹啉的苯并环之间存在π-π相互作用,实现了喹啉催化氢化的手性控制。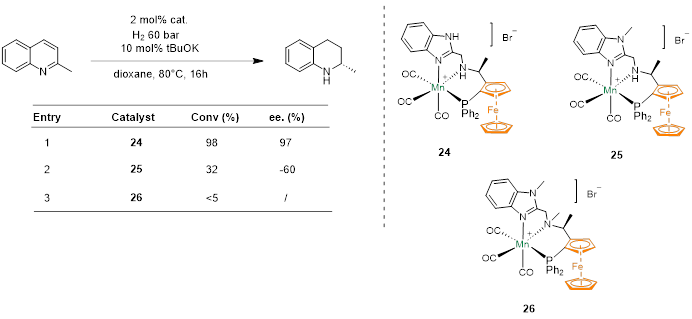
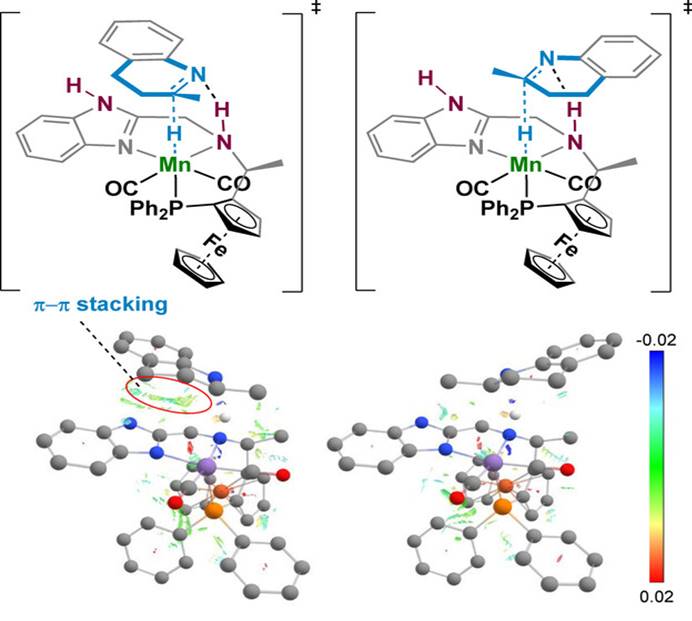
Fig. 7. Manganese-Catalyzed Asymmetric Hydrogenation of Quinolines Enabled by π–π Interaction
使用Noyori-Ikariya催化剂氢化3,3-二取代3-H吲哚时,生成金属氢物种的同时会生成等量的质子,使亚胺氮被质子化。质子化的亚胺活性比较高,很快就会被钌-氢物种氢化。如果底物中含有吡啶基团,生成的质子会转移到吡啶氮上,降低了底物的活性,这时使用Noyori-Ikariya型催化剂就难以实现对3,3-二取代3-H吲哚的氢化。而使用PNN-Mn催化剂时,因为锰-氢物种活性高,不需要将亚胺质子化就能实现底物的氢化。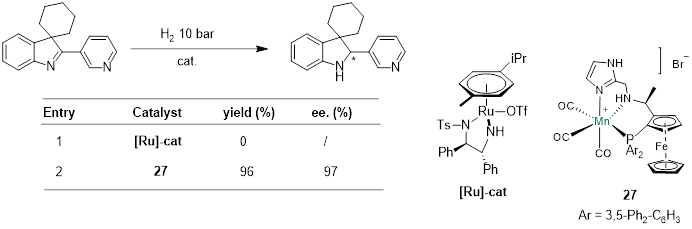
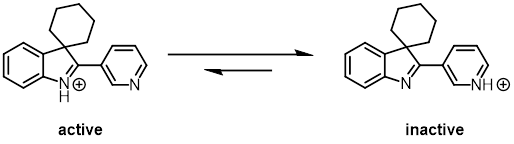
Fig. 8. Manganese-Catalyzed Asymmetric Hydrogenation of 3H-Indoles
刘强课题组发现使用PNN-Mn络合物进行喹喔啉不对称氢化时,通过控制反应条件可以实现分别得到顺式或反式的氢化产物,反应分两步将两个双键分别氢化,第二步氢化时催化剂进攻的面决定了产物的顺反。使用普通溶剂时钠离子在配体的咪唑环和底物的苯并环之间的阳离子-π相互作用起到了定位的功能,配体控制了另一个双键氢化的手性就得到了反式产物。而使用15-冠-5作为溶剂时,钠离子被束缚,阳离子-π作用消失,第二步双键氢化更倾向于从位阻小的一侧接近,从而生成顺式的产物。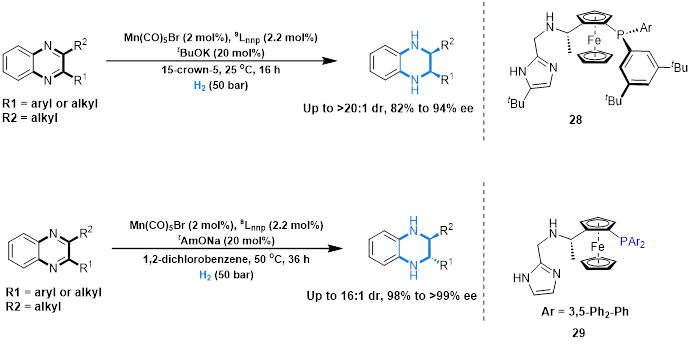
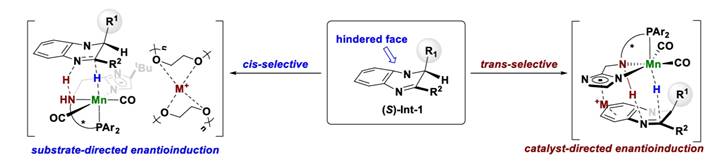
Fig. 9. Stereodivergent asymmetric hydrogenation of quinoxalines
近日,刘强课题组成功将σ-诱导活化作用引入了不对称氢化反应中,实现了在亚胺的不对称氢化中甲基乙基、以及比甲基乙基更难的丙基丁基的区分[15]。
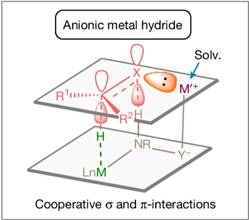
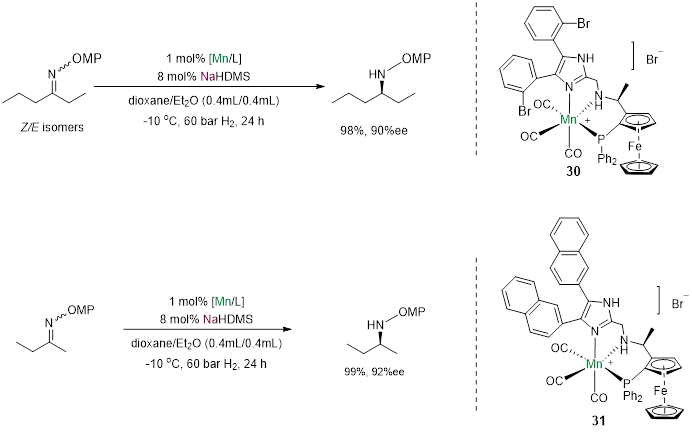
Fig. 10. Control experiments including sodium cation capture, the change of base and solvent
向体系中加入15-冠-5会使使ee值大幅降低,金属离子由钠换成钾也导致了ee值的降低,说明钠离子在手性控制中起到了极为重要的作用。

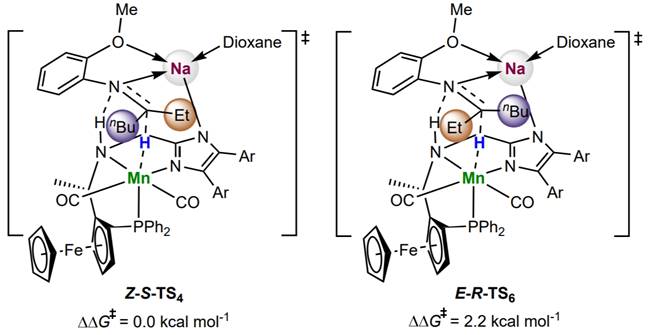
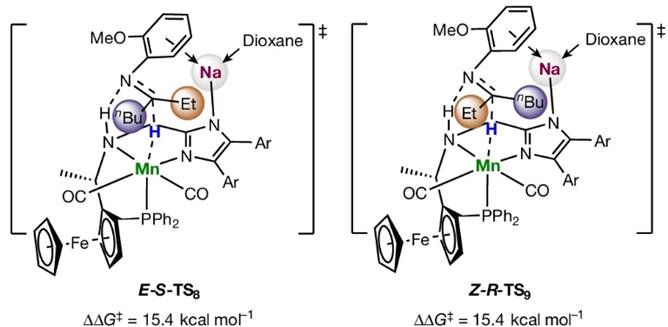
Fig. 11. Four hydride transfer transition states involving sodium cations. For hydride transfer transition states via σ-coordination interactions, Z-S-TS4 is favored over E-R-TS6 by 2.2 kcal mol-1; For hydride transfer transition states via cation-π interactions, the energy barriers via E-S-TS8 and Z-R-TS9 are 15.4 kcal mol-1 higher than that via the favorable transition state Z-S-TS4.
计算表明,钠离子与配体的咪唑基团并不处于同一平面内,而是略高于咪唑环的平面,从而实现了在底物氮在σ-键方向上的σ-诱导活化,除此以外,底物苯环上的甲氧基与钠离子的配位也对底物取向有重要的作用。同时配体氮上的氢与金属氢从π键的方向接近底物,实现了底物在π-键方向的活化。二氧六环与钠离子配位形成了巨大的位阻,让底物中较大的基团远离钠离子的方向。催化剂在亚胺C=N双键σ-轨道方向和π-轨道方向共同对底物作用,实现了强大的活化能力和立体控制能力。而当金属离子换成钾时,由于钾的半径更大,催化剂的立体控制能力随之降低,不对称氢化的立体选择性也随之下降。
总结
锰催化氢化虽然发展比较晚,但是近几年的发展也比较迅速,人们对机理的认识从最早简单的外球机理,到碱金属离子介导的π-轨道方向活化,再到σ-诱导活化,配体也从简单的消旋PNP配体逐步发展为具有强大立体选择性和催化活性的二茂铁PNN配体,均相锰催化不对称氢化反应在反应活性和不对称选择性上显示了巨大的潜力。
(非常感谢刘强教授课题组王明扬博士对本文的指导)
参考文献:
- [1] Saravanakumar Elangovan, Christoph Topf, Steffen Fischer, Haijun Jiao, Anke Spannenberg, Wolfgang Baumann, Ralf Ludwig, Kathrin Junge, and Matthias Beller*, Selective Catalytic Hydrogenations of Nitriles, Ketones, and Aldehydes by Well-Defined Manganese Pincer Complexes, J. Am. Chem. Soc. 2016, 138, 8809–8814.
- [2] Fabian Kallmeier, Torsten Irrgang, Thomas Dietel, Rhett Kempe*, Highly Active and Selective Manganese C=O Bond Hydrogenation Catalysts: The Importance of the Multidentate Ligand, the Ancillary Ligands, and the Oxidation State, Angew. Chem. Int. Ed. 2016, 55, 11806–11809.
- [3] Saravanakumar Elangovan, Marcel Garbe, Haijun Jiao, Anke Spannenberg, Kathrin Junge, Matthias Beller*, Hydrogenation of Esters to Alcohols Catalyzed by Defined Manganese Pincer Complexes, Angew. Chem. Int. Ed. 2016, 55, 15364–15368.
- [4] Marcel Garbe, Kathrin Junge, Svenja Walker, Zhihong Wei, Haijun Jiao, Anke Spannenberg, Stephan Bachmann, Michelangelo Scalone, Matthias Beller*, Manganese(I)-Catalyzed Enantioselective Hydrogenation of Ketones Using a Defined Chiral PNP Pincer Ligand, Angew. Chem. Int. Ed. 2017, 56, 11237–11241.
- [5] Magnus B. Widegren, Gavin J. Harkness, Alexandra M. Z. Slawin*, David B. Cordes*, Matthew L. Clarke*, A Highly Active Manganese Catalyst for Enantioselective Ketone and Ester Hydrogenation, Angew. Chem. Int. Ed. 2017, 56, 5825–5828.
- [6] M. Sc. Viktoriia Zubar, Yury Lebedev, Luis Miguel Azofra, Luigi Cavallo*, Osama El-Sepelgy*, Magnus Rueping*, Hydrogenation of CO2-Derived Carbonates and Polycarbonates to Methanol and Diols by Metal–Ligand Cooperative Manganese Catalysis, Angew. Chem. Int. Ed. 2018, 57, 13439.
- [7] Yujie Wang, Lei Zhu, Zhihui Shao, Gang Li, Yu Lan*, and Qiang Liu*, Unmasking the Ligand Effect in Manganese-Catalyzed Hydrogenation: Mechanistic Insight and Catalytic Application, J. Am. Chem. Soc. 2019, 141, 17337–17349.
- [8] Yujie Wang, Shihan Liu, Haobo Yang, Hengxu Li, Yu Lan*, and Qiang Liu*, Structure, reactivity and catalytic properties of manganese-hydride amidate complexes, Nat. Chem. 2022, 14, 1233–1241.
- [9] Shihan Liu, Haobo Yang, Ya-Nan Wang, Qiaoqiao Zhao, Yujie Wang*, Ruopeng Bai*, Qiang Liu*, and Yu Lan*, Enhancing Hydride Transfer in Catalytic Hydrogenation via σ-Electron-Induced Polarization of Imines, J. Am. Chem. Soc. 2024, 146, 24, 16357–16362.
- [10] Marcel Garbe, Kathrin Junge, Svenja Walker, Zhihong Wei, Haijun Jiao, Anke Spannenberg, Stephan Bachmann, Michelangelo Scalone, Matthias Beller*, Manganese(I)-Catalyzed Enantioselective Hydrogenation of Ketones Using a Defined Chiral PNP Pincer Ligand, Angew. Chem. Int. Ed. 2017, 56, 11237.
- [11] Linli Zhang, Yitian Tang, Zhaobin Han, Kuiling Ding*, Lutidine-Based Chiral Pincer Manganese Catalysts for Enantioselective Hydrogenation of Ketones, Angew. Chem. Int. Ed. 2019, 58, 4973–4977.
- [12] Chenguang Liu, Mingyang Wang, Shihan Liu, Yujie Wang, Yong Peng, Yu Lan*, Qiang Liu*, Manganese-Catalyzed Asymmetric Hydrogenation of Quinolines Enabled by π–π Interaction, Angew. Chem. Int. Ed. 2021, 60, 5108–5113.
- [13] Chenguang Liu, Mingyang Wang, Yihan Xu, Yibiao Li, Qiang Liu*, Manganese-Catalyzed Asymmetric Hydrogenation of 3H-Indoles, Angew. Chem. Int. Ed. 2022, 61, e202202814.
- [14] Chenguang Liu, Xufang Liu, Qiang Liu*, Chem 2023, 9, 2585–2600.
- [15] Mingyang Wang, Shihan Liu, Hao Liu, Yujie Wang, Yu Lan* and Qiang Liu*, Asymmetric hydrogenation of ketimines with minimally different alkyl groups, Nature 2024, 1–2.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
















No comments yet.