
作者:石油醚
引言
近日, Pfizer的科学家一条连续流的生产工艺,高效制备tert-Butyl 4-(6-Chloropyridin-2-yl)piperidine-1-carboxylate。
“Development of a Flow Process to tert-Butyl 4-(6-Chloropyridin-2-yl)piperidine-1-carboxylate
Aaron F. BaldwinNga M. DoXican HeCraig J. KnightTaegyo LeeBin LiuMark Duane McLawsPhilip PeachAngela L. A. Puchlopek-DermenciRachel RuestSteven J. R. Twiddle*Long YuanGuodu ZhangDavid F. Fernández*
Org. Process Res. Dev. 2026. DOI : 10.1021/acs.oprd.5c00467”
正文
活性药物成分(API)起始物料(SM)是用于合成 API 分子的关键化合物。Tert-butyl 4-(6-chloropyridin-2-yl)piperidine-1-carboxylate (1)是合成GLP-1激动剂Danuglipron的关键片段,用于Pfizer开发2型糖尿病及减肥治疗药物。该分子构成Danuglipron的核心骨架:吡啶环2位连有N-Boc保护的哌啶,便于氮原子后续修饰;6位为氯原子,支持该位点官能化。鉴于临床需求量大,因此需要一种能够提供多吨级产量的工艺。
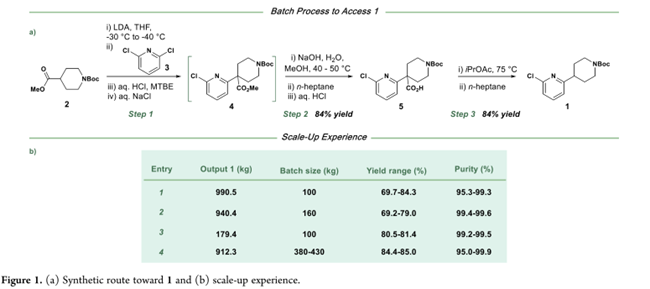
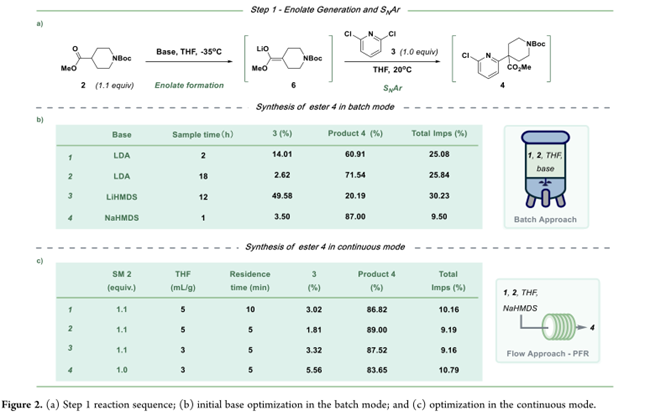
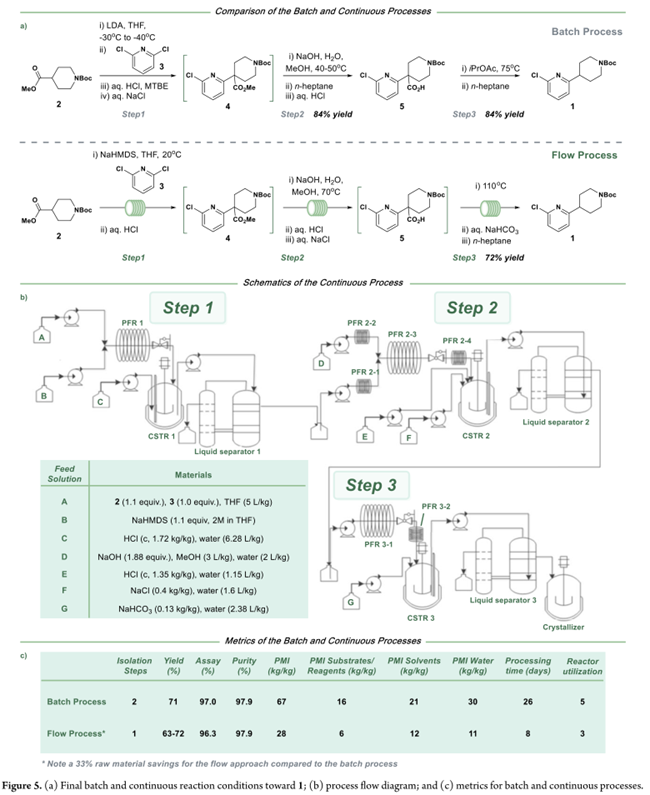
图1a是化合物1的合成路线和放大过程。即step 1:–30至–40 °C下,用LDA将N-Boc-4-哌啶甲酸酯(2)转化为烯醇盐,再与2,6-二氯吡啶(3)发生取代;Step 2:淬灭后不经分离,直接水解得羧酸5;Step 3:最后经脱羧得到目标物1。该批工艺优势明显:原料廉价易得,水解与脱羧使用常规试剂和工艺友好型溶剂,操作安全、简便,已实现100–400 kg/批次稳定生产(图1b)。但放大时存在三大瓶颈:(1)LDA需现配现用,低温下设备受限,制约产能;(2)向反应器投加2,6-二氯吡啶(3)前若未精准控温,易致反应停滞并产生杂质;(3)烯醇盐与3的反应缓慢,常需>15 h。因此,Pfizer的科学家聚焦于优化现有工艺。鉴于第一步对温度敏感且操作苛刻,优先探索连续流的方案:拟在15–25 °C下连续生成烯醇盐并即时与3反应,以提升过程可控性、产率与产品质量。随着需求增长,连续工艺在效率、质量一致性及可持续性(如缩短周期、降低能耗与废液量)方面的优势将更为突出。
首先,连续工艺开发初期,采用2-甲基四氢呋喃(2-MeTHF)开展快速筛选,旨在实现step 1至Step 3的单溶剂串联反应。结果表明:步骤1在2-MeTHF中反应性能良好,且动力学特性适配流动体系,据此推进后续开发。
step 1 包含两个主要阶段:1)N-Boc-4-哌啶甲酸酯(2)在碱作用下生成烯醇盐中间体6;2)6与2,6-二氯吡啶(3)发生SNAr反应得产物4。初始选用LDA(Figure 2b,entry 1),因强碱性且匹配既往批次条件,但反应缓慢:2 h仅得61%产物4,残留14%原料3;18 h后转化率仅升至72%,并产生多种未鉴定杂质。对比评估其他强碱后,NaHMDS表现最优——反应更快、转化率更高、杂质更少(Figure 2b,entry 4),遂选定为后续开发用碱。
为了实现温和条件下的单步(step 1)连续化,采用THF溶液体系,在20 °C下将2、3与NaHMDS溶液混合,可快速完成烯醇化及取代反应。管式平推流反应器(PFR)被确定为最适反应平台,并系统考察了关键工艺参数。结果表明:停留时间5–10 min、浓度3 L/kg(较5 L/kg提升通量并降低过程质量强度PMI)、NaHMDS与2均为1.1 equiv、3为1.0 equiv、温度20 °C为最优组合(图2c)。其中,2过量0.1 equiv用于补偿NaHMDS活性波动、烯醇化效率及微量水干扰。此外,为防止中间体4在水解前脱保护,设计连续淬灭与相分离工艺:间歇工艺中需盐酸淬灭→TBME萃取→多次洗涤→蒸馏浓缩;而连续工艺简化为——盐酸中和至pH 7–8后直接分液,有机相直接进入下一步水解。
Step 2为甲酯4的水解,生成羧酸5,以备后续脱羧。设计目标是:在step 1有机相经简单淬灭与分液后,直接进行水解,且该水解不受THF体系中微量水的影响。
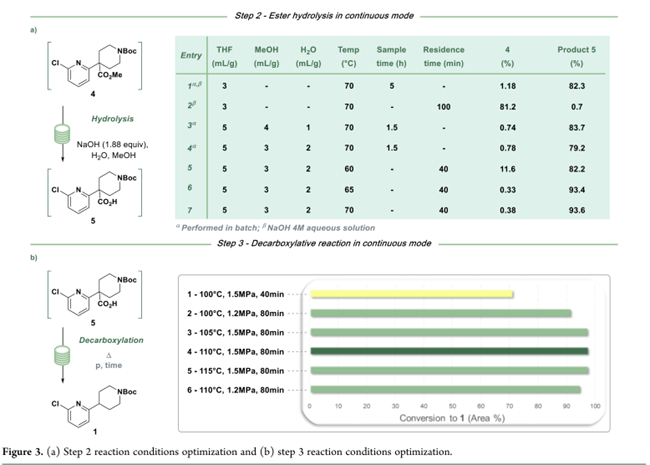
间歇工艺长期采用成本低、易得的NaOH(1.88 equiv,4 M),以2,6-二氯吡啶(3)为计量基准;70 °C下反应可实现高转化率(图3a,entry 1)。但该混合物不均一,在管式反应器中易堵塞并降低转化率(Figure 3a,entry 2)。为改善均一性,考察甲醇、额外水及THF稀释的影响;最终确定3 L/kg甲醇 + 2 L/kg水的组合,可确保反应与淬灭全程均相(图3a,entry 4),并在≥65 °C下实现羧酸5的完全转化(图3a,entries 6–7)。连续工艺中,NaOH溶于甲醇/水混合液,与含4的步骤1流出液在线混合;随后进入CSTR,用盐酸水溶液淬灭,并加入NaCl强化相分离;所得含5的有机相直接用于步骤3脱羧。
Step 3脱羧原在间歇工艺中使用iPrOAc作溶剂(起始物料为分离所得酸5)。为规避连续流程中低效的溶剂切换,直接沿用步骤2的甲醇/THF体系。Figure 3b显示:该体系在宽温度范围内稳健,最终选定110 °C、80 min停留时间、1.5 MPa反应压力(Figure 3b,entry 4)。
化合物1的最终分离沿用原工艺:步骤3反应液经NaHCO₃水溶液在CSTR中淬灭,分液后,有机相减压置换为过饱和正庚烷溶液,实现可控结晶,再经过滤、洗涤得纯品。
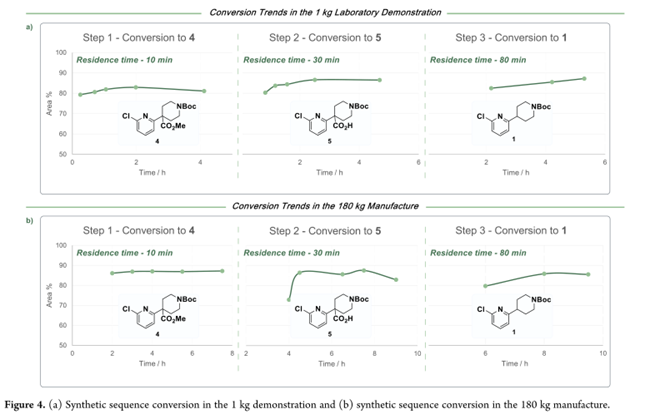
经系统优化,确立三步串联连续工艺:第一步以NaHMDS为碱,在20 °C下使N-Boc-哌啶-4-甲酸酯(2)烯醇化,并与2,6-二氯吡啶(3)反应10 min;淬灭分液后,有机相直接进入第二步——70 °C下与NaOH/甲醇水溶液在管式反应器中皂化30 min(转化率85%);随后于CSTR中用盐酸酸化并加入20% NaCl水溶液促相分离,得羧酸5;第三步为110 °C管式反应器中热脱羧。最终,有机相经正庚烷溶剂置换、结晶、过滤及干燥,获得化合物1。实验室完成1 kg示范实验(Figure 4a),三步转化率高度稳定,收率72%,纯度>98%。后续开展180 kg级生产:投料142.2 kg原料3,全程中间体4转化率稳定在87%(Figure 4b)。实际得188.5 kg产物1;收率略低于理论值,主因是系统达到稳态所需时间占比较高。Figure 5c显示:连续工艺总体PMI降至28 kg/kg,不足间歇工艺(67 kg/kg)的50%,且原材料、溶剂与水耗显著降低。
结论
化合物1连续合成工艺的开发基于三大因素:强化低温步骤的过程精细控制、提升可持续性、缩短制造周期——三者均随产能扩大而愈发关键。该工艺已成功由间歇模式转为连续模式,并完成1 kg与188 kg两级验证。产率与纯度与原批工艺相当。连续工艺使PMI降低超50%,总制造时间压缩至批工艺的30%,主要得益于取消中间体隔离及反应/后处理单元操作的集成优化。本文证实,连续制造可带来切实可观的工艺升级效益。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/



















No comments yet.