
作者:石油醚
引言
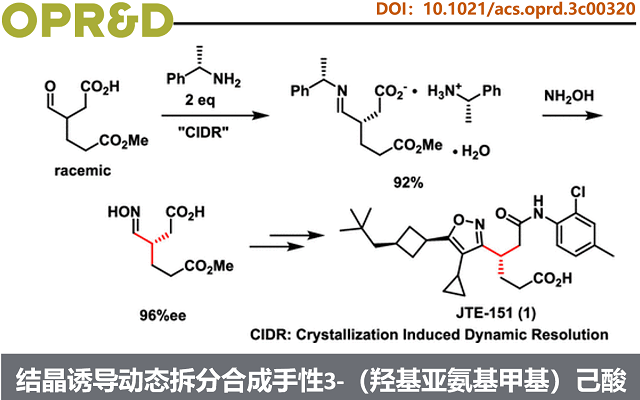
近日,Japan Tobacco Inc.基于结晶诱导动态拆分(CIDR),报道了一种六步合成手性3-(羟基亚氨基)己二酸4的实用路线。该中间体经1,3-偶极环加成构建异噁唑环,再经Ag2O催化脱羧,直接得到异噁唑23——ROR-γ抑制剂JTE-151的API起始物料。
Practical Synthesis of Chiral 3‑(Hydroxyiminomethyl)adipic Acid, a Key Intermediate of the ROR‑γ Inhibitor JTE-151, via CrystallizationInduced Dynamic Resolution.
Hiromu Takiguchi,* Takashi Watanabe, Takashi Ogo, Hideaki Ishibashi, Katsuyuki Yokota, Shingo Obika, and Takashi Inaba*
Org. Process Res. Dev. 2023, 27, 2355–2364. Doi: 10.1021/acs.oprd.3c00320
正文
RORγ 是 Th17 细胞分化、存活和增殖的关键调控因子,主要通过上调 IL-23 受体实现。Th17 细胞分泌 IL-17A、IL-17F、IL-21 和 IL-22,驱动银屑病、类风湿关节炎、多发性硬化症和炎症性肠病等多种难治性自身免疫疾病。因此,RORγ 已成为重要药物靶点,全球制药企业正积极开发高活性、临床可行的 RORγ 抑制剂。
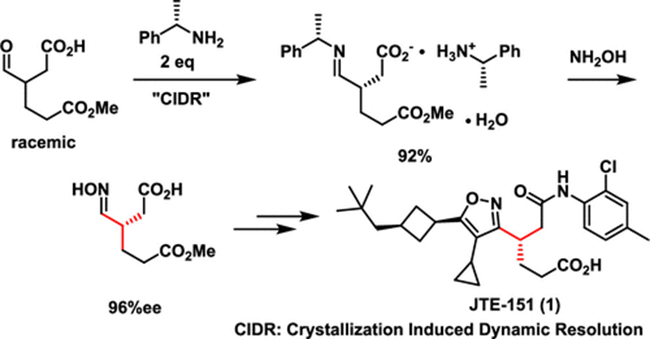
近期,科学家发现了小分子 RORγ 抑制剂 JTE-151(1),并已推进至临床试验阶段。其中JTE-151是以异噁唑作为骨架核心,并在异噁唑的3-, 4-, 5-位含有手性己二酸单酰胺、环丙基和cis-3-新戊基环丁基(scheme 1)。目前,以采用1,3-偶极环加成作为主要策略合成此类化合物。即以β-酮酯3为亲合偶极体,与原位生成的手性腈氧化物(源自肟4)反应,可高区域选择性地构建5-位含3-新戊基环丁基、4-位含酯基的异噁唑骨架;随后通过脱羧、卤化及Suzuki–Miyaura偶联(环丙基硼酸),将4-位酯转化为环丙基。肟4含两个不同保护的羧基及邻近肟基的手性中心,合成难度大。近日,Japan Tobacco Inc.基于结晶诱导动态拆分(CIDR),报道了一种六步合成手性3-(羟基亚氨基)己二酸4的实用路线。该中间体经1,3-偶极环加成构建异噁唑环,再经Ag2O催化脱羧,直接得到异噁唑23——ROR-γ抑制剂JTE-151的API起始物料。
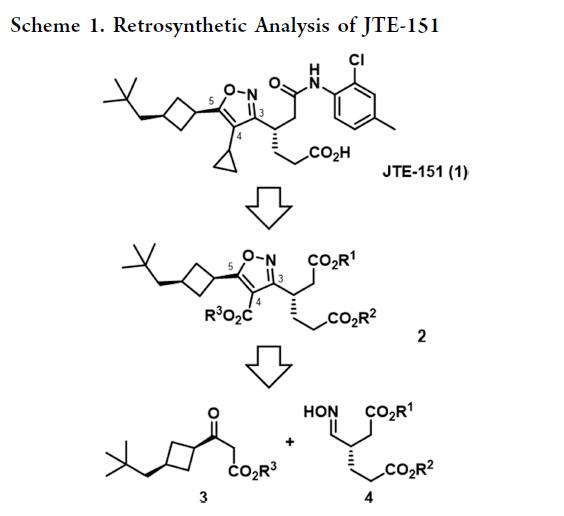
首先,作者对三种合成关键手性中间体(S)-4a(3-(羟基亚氨基甲基)己二酸)及其等效物(7)(scheme 2)的策略进行了评估:不对称烷基化、不对称氢化和结晶诱导动态拆分(CIDR)。
不对称烷基化,以化合物5为底物,在−78 °C下用NaHMDS作碱,与叔丁基溴乙酸酯反应,借助Evans手性噁唑烷酮辅基,以>95% de获得6。该路线适用于药物发现及早期开发,但后续需多步还原6中的噁唑烷酮酰胺为醛以制备7;且7中苄基保护的羟基须在异噁唑环构建完成后氧化为受保护羧基。
进入中后期开发后,亟需更简短、经济的(S)-4合成路线,避免低温操作与色谱纯化。因此,作者转向催化不对称氢化与CIDR,并设计底物含两个差异化羧基及一个肟基或醛基,从而全程规避氧化/还原步骤。据此,α,β-不饱和酸8(用于氢化)和醛9(用于CIDR)被选定为目标底物,二者均源自共同前体13(scheme 3)。
13经改进文献法两步合成:首先,醛11(由10经 Rosenmund还原制得)、乙醛酸与吗啉发生Mannich反应;随后,中间体12在酸性条件下一锅完成双吗啉基团的β-消除与水解,以65%收率得到结晶性13。13与羟胺直接反应,高收率(95%)、高几何纯度地生成氢化底物8;而经Rh催化氢化,则得到醛9与半缩醛的平衡混合物(DMSO-d₆中比例23:77)。
尽管α,β-不饱和羧酸的不对称氢化已有诸多成功报道,但底物8中肟基在还原条件下的耐受性及对对映选择性的影响此前未知。我们基于大规模可获得性,筛选了多种金属/手性配体组合,发现铑体系在保持肟基完整的同时可实现良好转化。
然而,所有体系均未能达到工业化所需的对映体过量(ee)。最优结果为[Rh(cod)₂]BF₄/(R)-CyBINAP(S/C = 300),仅得(R)-4a(目标(S)-对映体的反向异构体)46% ee。此外,8稳定性差,不适用于放大生产。鉴于上述局限,转向9的结晶诱导动态拆分(CIDR)。
CIDR是工业化制备手性化合物的理想策略:无需低温、高压设备或昂贵试剂;通常单步操作、条件温和、官能团兼容性优异,可显著缩短合成路线。已有报道利用手性伯胺与α-差向异构醛形成亚胺,再通过选择性结晶富集单一立体异构体实现CIDR。而化合物9兼具α-差向异构醛基和游离羧基,因而具备三类CIDR路径:(a)仅醛基成亚胺结晶;(b)仅羧基成盐结晶(醛基保留);(c)醛基与羧基协同形成亚胺盐结晶。市售手性胺筛选确认,(S)-1-苯乙胺可与9形成稳定结晶复合物。
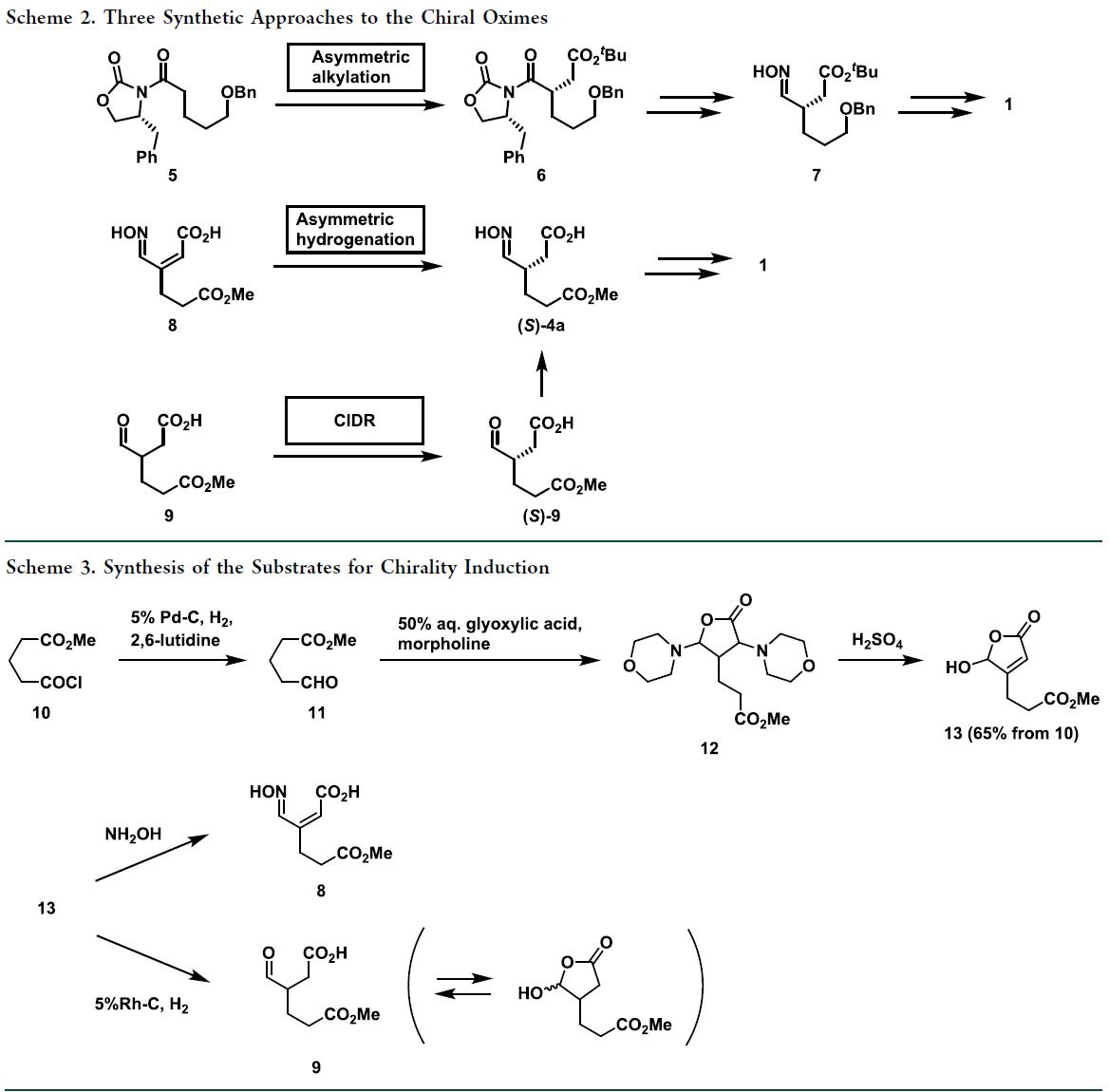
其次,9与等量(S)-1-苯乙胺(14)在室温下于己烷/乙酸乙酯中混合,直接析出γ-氨基-γ-丁内酯15的单一晶型晶体,产率66%(scheme 4)。单晶X射线分析确认其结构(Figure 1),且14的手性同步控制15中β-与γ-位两个立体中心。然而,15经盐酸羟胺室温处理后,虽1H NMR显示其为非对映体纯,却仅以91% ee得到非目标(R)-4a,表明转化过程中发生部分消旋。改用2 equiv 14进行CIDR,则高产率(92%)获得新型晶体16。X射线证实其为亚胺盐结构(Figure 1),且经硫酸羟胺处理,可在70 g规模以96% ee直接制得目标(S)-4a。值得注意的是:仅通过调控14的用量(1 vs. 2 equiv),即可选择性获得(R)-或(S)-4a——同一手性胺实现构型翻转。该转化需将固体16分批加入略过量的硫酸羟胺水/乙腈溶液;反向加料会导致光学纯度显著下降。反应完成后,加入等量硫酸,使14以硫酸盐形式定量沉淀。过滤即回收>95%的14(以硫酸盐计)。
最后,17 至 JTE-151 API 起始物料(23)的转化包含两个关键步骤(scheme 5):(1)与β-酮酯3a(由19以89%收率制得,无异构化)发生1,3-偶极环加成,构建异噁唑核心;(2)选择性脱除异噁唑4位引入的羧基。该环加成采用一锅法(Table 1):17经NCS氯化生成氯肟18,随即在t-BuOK/3a作用下原位产生腈氧化物,并被3a烯醇盐高效捕获,直接形成异噁唑。粗产物2a经Pd/C氢解脱除苄基与4-硝基苄基,所得二酸20以环己胺盐21形式结晶纯化(收率62%,以16计)。脱羧步骤采用Gooßen条件(5 mol% Ag₂O、10 mol% K₂CO₃,NMP,135 °C),实现20高选择性转化为单酸22。22经SOCl₂转化为酰氯后,与t-BuOH酯化得叔丁酯23——JTE-151的API起始物料候选物。粗品23经活性炭脱色、水/2-丙醇重结晶,获得高纯度产物(HPLC纯度98.99%,ee 98.5%),收率86%(以21计)。 23的纯度与前期通过不对称烷基化路线(scheme 2)制备的23相当;后者已成功用于36 kg级JTE-151临床样品合成,总收率75%(scheme 6)。
23至JTE-151的转化路径成熟可靠:(i)23经NBS溴化得24,再与环丙基硼酸在PdCl₂(amphos)₂催化下Suzuki-Miyaura偶联得25;(ii)25在酸性条件下选择性脱叔丁基酯,所得26以(S)-1-苯乙基苄胺盐形式纯化;(iii)26经酰氯化、与2-氯-4-甲基苯胺缩合得27,再碱性水解甲酯即得JTE-151。
结论
综上,Japan Tobacco Inc.开发一条高实用性路线,通过消旋醛9与2 equiv (S)-1-苯乙胺(14)的结晶诱导动态拆分(CIDR),高效合成手性3-(羟基亚氨基甲基)己二酸(S)-4a。该CIDR工艺总收率92%(含消旋体9的制备),在70 g规模下,以96%ee获得(S)-4a对映体,且14回收率>95%。由此所得(S)-4a经五步转化(含1,3-偶极环加成构建异噁唑环及Ag₂O催化的4-位羧基脱羧)即得JTE-151的API起始物料23。该CIDR路线显著降低JTE-151的生产成本。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/

















No comments yet.