
作者:王辉
导读:
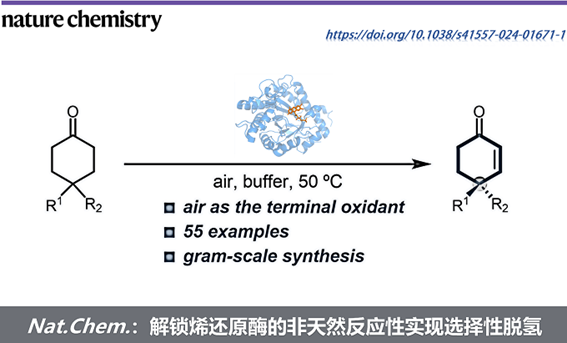
近日,西湖大学叶宇轩课题组在Nature Chemistry上发表了题为「Unmasking the Reverse Catalytic Activity of ‘Ene’-Reductases for Asymmetric Carbonyl Desaturation」的研究论文。该小组从一类底物范围好、热稳定性高、易于表达的烯还原酶出发,成功把其从还原酶改造成去饱和化酶,建立了一个通用的酶催化羰基去饱和化反应平台,合成了一系列含有远端四级手性中心的高价值环己烯酮产物。此酶催化反应体系条件温和、操作简单、易于放大,展现出与已有的化学合成方法互补的反应性和选择性
“Unmasking the Reverse Catalytic Activity of ‘Ene’-Reductases for Asymmetric Carbonyl Desaturation
Hui Wang, Bin Gao, Heli Cheng, Shixuan Cao, Xinyi Ma, Yinjuan Chen, Yuxuan Ye*
Nat.Chem. 2024, ASAP. DOI: https://doi.org/10.1038/s41557-024-01671-1”

正文:
α,β-不饱和羰基化合物在制药、农药和材料等多个领域具有重要的作用。在预先构建的分子框架中直接脱氢引入与羰基相邻的C=C双键是构建α,β-不饱和羰基化合物的理想策略之一。近几十年来,合成化学家发展了许多催化体系来实现此过程。例如羰基α位卤化-消除的串联过程;Saegusa-Ito氧化;高价碘氧化;新型氧化剂 (SeO2、N-叔丁基苯基亚磺酰亚胺氯、IBX和AZADO等) 介导的直接脱氢;过渡金属催化;电催化等 (图1a)。然而,目前已有的方法要么需要多步合成,要么需要在较强氧化性条件下进行,或使用较高当量的金属催化剂。更重要的是,目前还没有一个普适的催化体系,可以在去饱和化过程中实现立体化学的精确控制。这些局限都限制了直接脱氢去饱和化反应在合成化学中更广泛的应用。
近年来,酶催化剂因其选择性控制精确、易于改造进化和绿色环保等独特优势,越来越受到合成化学家的关注。自然界中存在一些天然催化羰基去饱和化反应的酶,但它们的底物普适性差,难以作为催化剂被广泛应用。为了克服这些局限,西湖大学叶宇轩课题组从一类底物范围好、热稳定性高、易于表达的烯还原酶出发,成功把其从还原酶改造成去饱和化酶 (图1b),建立了一个通用的酶催化羰基去饱和化反应平台,合成了一系列含有远端四级手性中心的高价值环己烯酮产物 (图1c)。此酶催化反应体系条件温和、操作简单、易于放大,展现出与已有的化学合成方法互补的反应性和选择性。相关成果发表于Nature Chemistry上。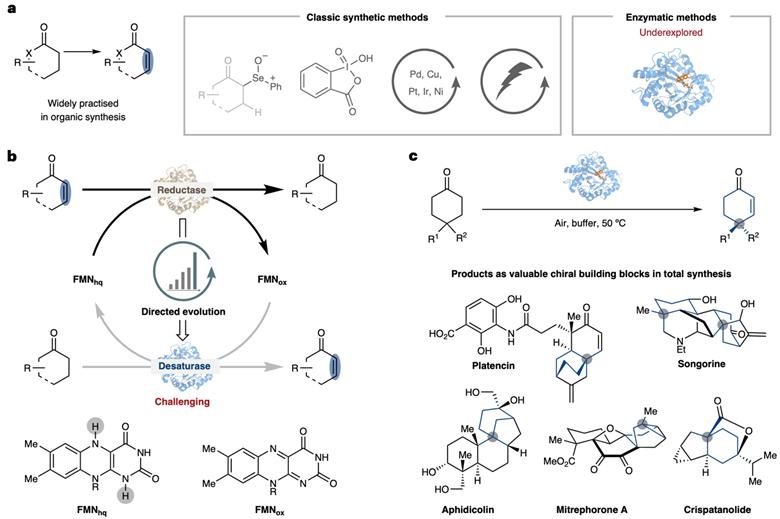
图1. 酶催化羰基去饱和化反应。图片来源:Nat. Chem.
作者首先建立了结构多样的烯还原酶库,以4-甲基-4-苯基-环己酮1作为模型底物,在氧气存在的条件下对ERED的脱氢反应性进行评估,发现CrS和GkOYE表现出较好的活性和对映选择性控制。当温度升高时,去饱和化反应性提高 (图2a)。随后作者以野生型CrS和GkOYE为起点,利用定向进化技术提高酶催化剂的反应效率。单突变体CrS1 (Y27L)、CrS2 (Y27C) 和CrS3 (Y27T) 分别以79% (98:2 er), 88% (98:2 er)和81% (97:2 er) 得到产物2;GkOYE-WT经过三轮进化得到一个三重突变体GkOYE3 (D73G、C26S、Y28V),可以以很高的效率催化2的生成 (96%,>99:1 er) (图2b)。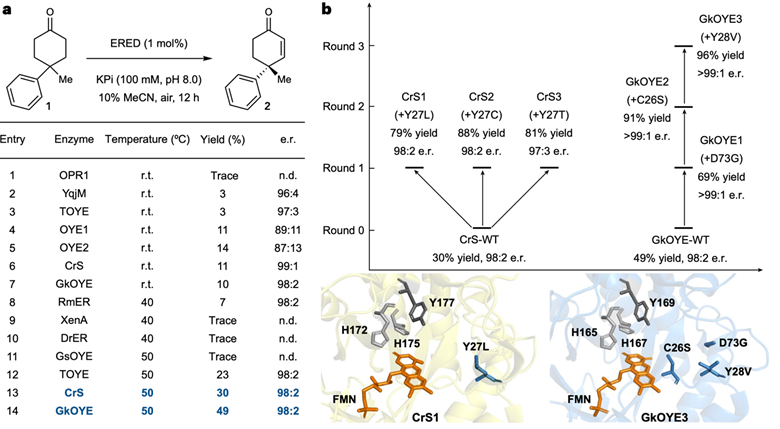
图2. 反应优化和定向进化。图片来源:Nat. Chem.
接着,作者收集了在定向进化中获得的有益突变体,建立了去饱和化酶库,并对反应的底物范围进行了探索。作者首先测试了多种4-烷基-4-芳基取代的环己酮底物,均具有良好的耐受性 (图3)。当使用含2-氨基苯基的环己酮作为起始原料8时,以99% (>99:1 er, >20:1 dr) 的收率得到环化的产物二氢吲哚酮9。值得注意的是,这种生物催化去饱和化方法对胺和硫醚等氧化敏感官能团表现出高的兼容性。此外,缺电子和富电子杂环都是能够在该反应条件下被兼容。除甲基外的各种烷基取代基底物也可以实现高立体选择性的脱氢。从相应的3-羟丙基底物18出发时,可以99%收率 (>99:1 er,>20:1 dr) 得到环化产物八氢色烯-7-酮19。此外,螺环底物 (20–23) 也能很好地转化。然而,4-羟基苯基的环己酮24不能被酶催化体系兼容,可能是由于该底物是酶的抑制剂。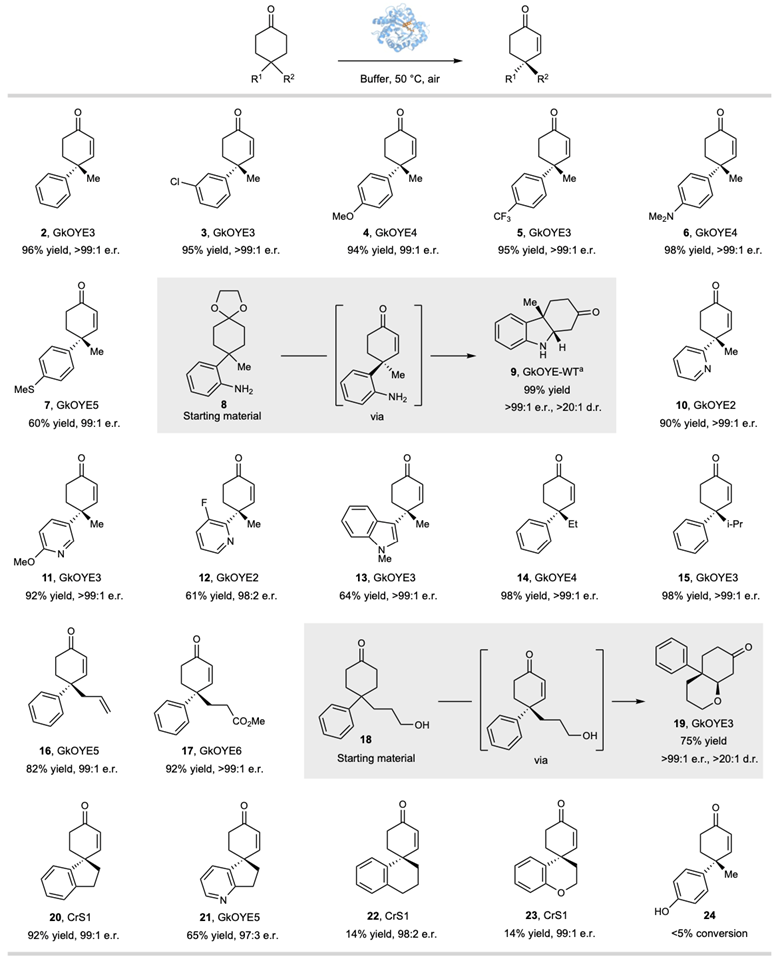
图3. 4-烷基-4-芳基环己酮去对称化脱氢反应。图片来源:Nat. Chem.
无需进一步的酶工程改造,此去饱和化方法可以被扩展到具有其它取代基团的环己酮底物。二烷基取代的环己烯酮25–33可以被高效去饱和化。对于含有甲基和乙基这两个差别很小的取代基的底物34,在没有经过定向进化的情况下仍然能以62:38 er得到目标产物。各类4-羟基-环己酮底物和4-酯基-环己酮底物都可以顺利的发生反应。此外,甲氧基51、乙酰氧基52和氰基53取代的环己酮也能被酶兼容。单取代的环己酮也能成功地实现去饱和化,以中等至良好的收率和高水平的对映选择性得到相应的产物54-58。4-羟基-4-炔基环己酮59在该条件下无法反应 (图4)。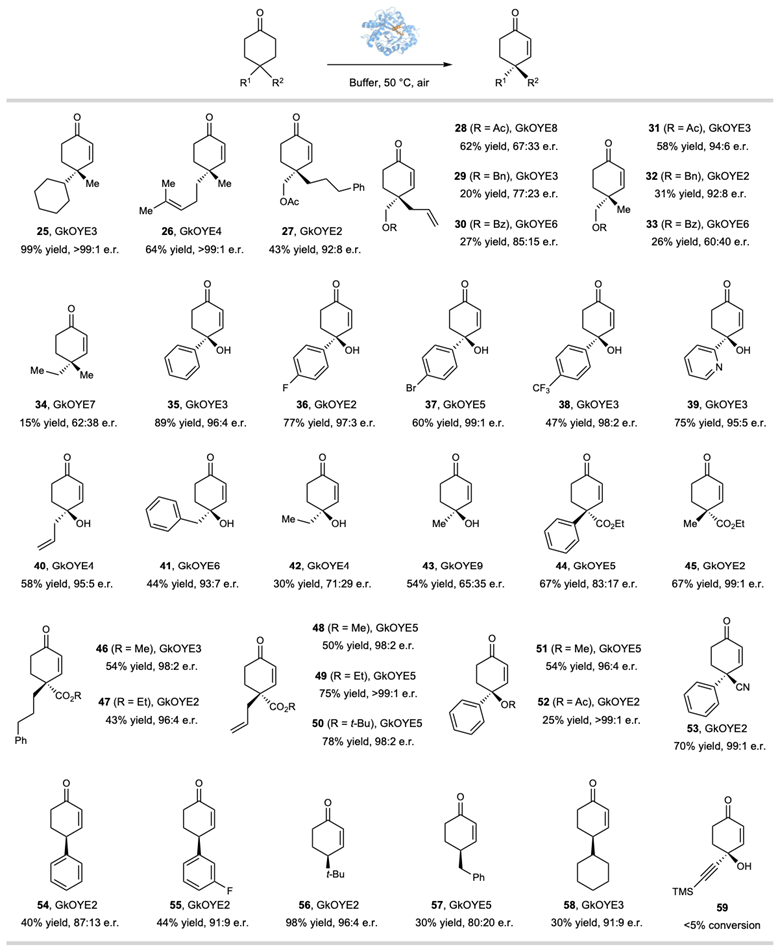
图4. 其它类别环己酮的去对称化脱氢反应。图片来源:Nat. Chem.
此酶催化反应体系条件温和,操作简单,可以很容易进行放大。作者首先对模型反应进行了克级规模放大:使用细胞裂解液可以88% 的产率,>99:1 er获得产物2,使用冻干裂解液可以81% 的产率、>99:1 er实现反应 (图5a)。此外,作者以克级规模高效制备了 (+)-Mesembrine,一种具有血清素再摄取抑制活性的生物碱。通过酶促去饱和化过程制备的环己烯酮产物2是应用广泛的合成前体,能够高立体特异性的转化为有价值的手性化合物62,或是经历底物控制的非对映选择性Diel-Alder反应,生成了带有三个相邻立体中心的六氢萘酮63 (图5c)。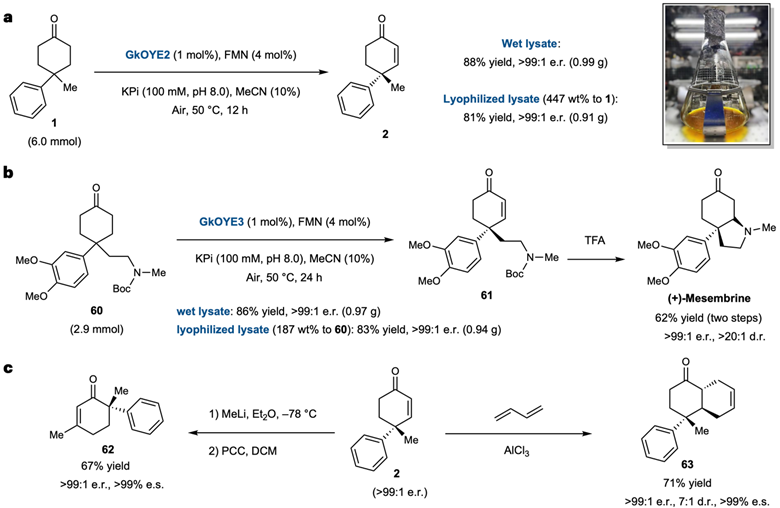
图5. 克级规模放大和衍生化。图片来源:Nat. Chem.
最后,作者对这一反应的机理进行了探索。此脱氢反应的可能机理是 (图6a): 底物在酶的活性空腔结合后,保守的酪氨酸残基阴离子 (GkOYE3的Y169) 对底物进行α-去质子化,形成烯醇中间体Int1。随后,一种反应机理是经历路径A,Int1将β-氢负转移至FMNox,得到去饱和化的烯酮产物2和FMNhq。此反应途径与ERED催化的天然还原过程相反,符合微观可逆性原理。另外一种反应机理是经历路径B,Int1与FMNox进行单电子转移 (SET),生成相应的α-酰基中间体Int2和黄素半醌FMNsq,然后进行质子转移 (PT) 和另一个单电子转移 (SET) 成2和FMNhq。此反应途径与近期报道的ERED催化的单电子氧化反应类似。最后,在酶催化剂释放产物2,FMNhq与空气中的分子氧发生反应,经历黄素氢过氧化物中间体,形成过氧化氢并重新生成氧化态辅因子FMNox。
基于这个机理模型,作者首先利用氢氘交换实验 (图6b) 和基因敲除实验 (图6c) 证明了Y169氨基酸残基对去饱和化反应的重要作用。自由基钟实验结果与β-氢负转移机制一致 (图6d)。接着,作者对该反应独特的β-氢负转移步骤进行了动力学研究,证明了反应的决速步是β-氢负转移。α-和β-位的动力学同位素效应 (KIE) 与所提出的α-去质子化和β-氢负转移的反应途径一致 (图6e)。动力学实验表明,定向进化后的突变体GkOYE3的催化去饱和化半反应速度比GkOYE-WT快约57倍,还原反应速度变慢约0.7倍,与氧反应速度比GkOYE-WT快约1.5倍。总体而言定向进化产生的突变体在所有三个关键基元步骤中都更适合去饱和化 (图6f)。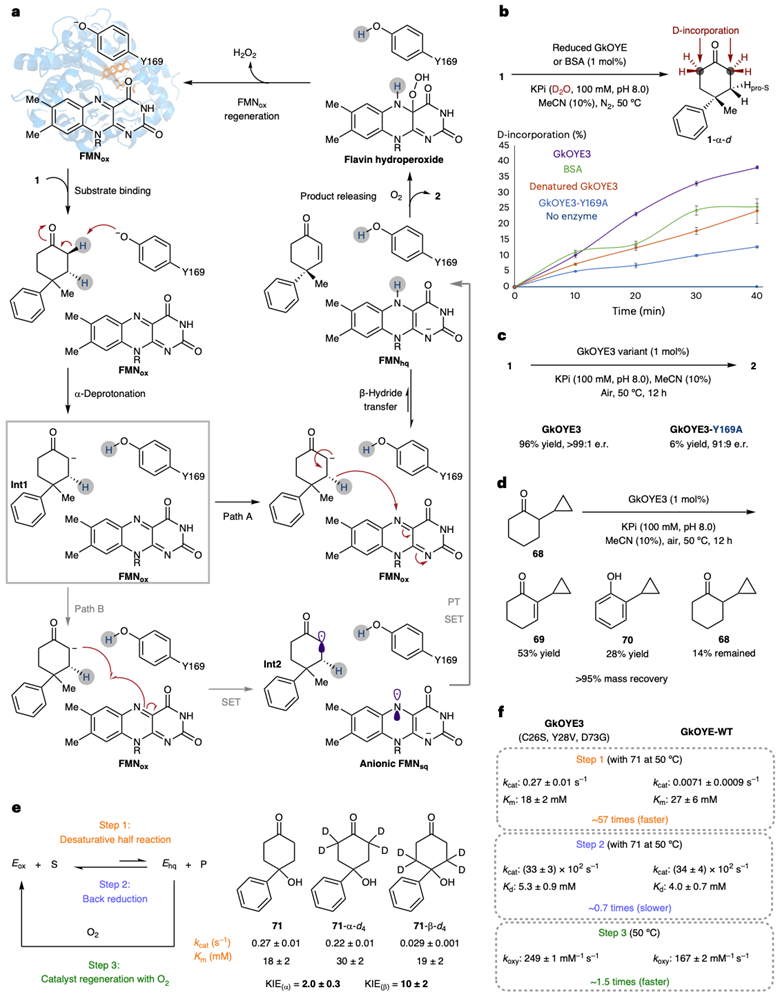
图6.反应机理;氢/氘交换实验;动力学实验。图片来源:Nat. Chem.
总结
叶宇轩课题组成功的解锁了烯还原酶的全新非天然去饱和化反应性,把它们从还原酶改造成了去饱和化酶。此创新生物催化去饱和化平台展现出与已有方法互补的反应性和优越的选择性,具有较好应用潜力。作者通过系统的机理研究,加深对酶催化去饱和化机理的理解,为此反应体系的进一步发展奠定了基础。
通讯作者简介

叶宇轩博士2013年于北京大学化学与分子工程学院获得学士学位,指导教师为王剑波教授和张艳教授。2018年在美国麻省理工学院获得博士学位,指导教师为Stephen Buchwald教授。2019–2022年在美国普林斯顿大学和康奈尔大学开展博士后研究,合作教师为Todd Hyster教授。叶宇轩博士于2022年9月全职加入西湖大学理学院担任特聘研究员,主要研究方向为新型酶催化非天然反应的发展和应用。叶宇轩课题组现面向国内外招聘博士研究生、博士后、科研助理,欢迎对酶催化感兴趣的同学申请。课题组主页:ye.lab.westlake.edu.cn。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/













No comments yet.