
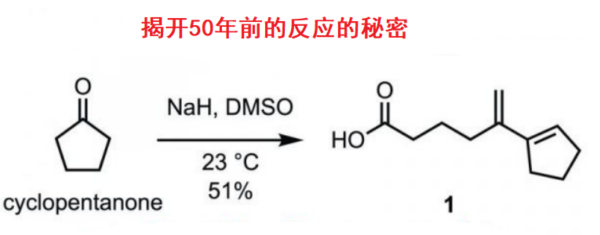
背景介绍
近年来通过过渡金属催化的C-H活化是一个十分热的方向,而小编其实也在从事这一方向,不过小编做的东西有一点小众-设计一款通过氢键控制的手性羧酸铑催化剂,并且通过配体与底物之间的氢键作用实现site,stereoselective的C-H插入反应。可能很多人对这领域不是特别了解,今天小编就在这里介绍一下羧酸铑催化的卡宾的插入反应。
催化循环
首先从催化循环机理(Scheme 1)走起。卡宾的插入基本都是以重氮为原料,该原料作为一个亲核试剂进攻羧酸铑催化剂I,形成中间体complex II,然后再脱氮气形成卡宾中间体III,这一脱氮气形成卡宾的步骤被认为是该反应的rate-determination step1,2,但是这只是相对于从I形成II而言,而且这跟催化剂的配体,重氮原料,分子内or分子间反应也是相关的,所以到底是不是还有待进一步的证据,到目前还没有一个切实的定论,至少小编是这么认为的。在形成卡宾活性中间体III后,与含有C-H键的原料进行C-H插入反应,这一步特别重要,也是探索了好多年,直到2002年,Nakayama group3通过计算给出了一个最有可能的推测-通过concerted mechanism进行反应,完成整个催化循环。
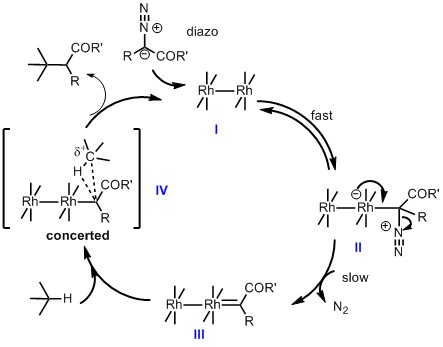
Scheme 1. Mechanism of rhodium-catalyzed carbene formation from Diazo Compounds
卡宾朝向
对于Scheme 1中形成的卡宾中间体III来说,卡宾相对于羧酸铑的O-Rh-O的平面的朝向问题对于手性催化也是非常重要的,很明显有两种可能(Figure 1),一种就是与O-Rh-O平行(eclipsed form),而另一种就是交叉构型(staggered form)。从立体位阻来看,很明显交叉构型的位阻(EDG:electron donating group or EWG: electron withdrawing group与配体取代基R之间的位阻)是相比于平行构型更小,应该是可能性大,其实不仅仅只是这个原因,最主要的是Rh的4d轨道与卡宾C的2p空轨道有个轨道的给电子交叠,从而有一个稳定卡宾中间体的效果,小编认为这才是主要原因,因此富电子的配体形成的卡宾更稳定,但是C-H插入的反应性会相对差一点。但是这也仅仅是一个推测,实验学科需要拿出确实的证据才能定论。
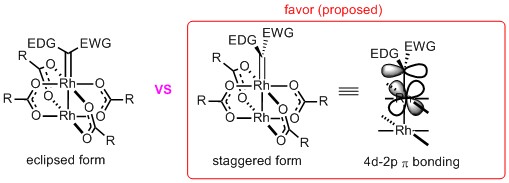
Figure 1. Proposed orientation of carbene ligand to oxygen ligand
证据在这里!
直到今年,Alois Furstner4等人拿到了卡宾中间体的单晶(Figure 2),从这个单晶也证实了确实卡宾的两侧取代基是与O-Rh-O这条线交叉配置的,并且,EWG部分(下图中的左侧酯基)是“躺着的”-平行于羧酸铑平面的,而右侧的EDG部分则是“站着的”-交叉于羧酸铑平面。从轨道很容易解释这个问题,右侧EDG这样的配置很明显导致了苯环的π轨道电子与卡宾的C1的2p轨道有一个共轭作用,从而使得该中间体更稳定。对于这个研究小编只能默默地点赞!
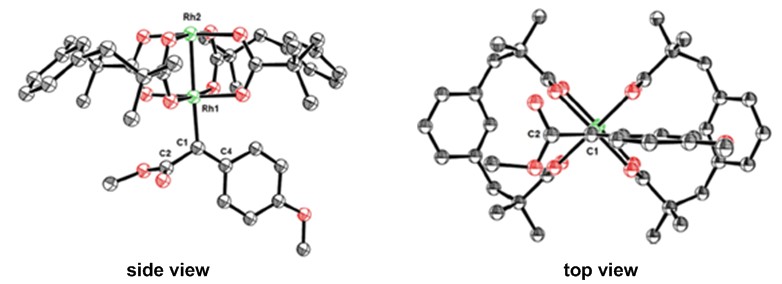
Figure 2. structure of donor/acceptor dirhodium{Rh2(esp)4} carbene in the solid state
具有代表性的手性羧酸铑催化剂
近年来手性羧酸铑催化剂的开发与应用发展的非常迅速,配体的不同对于整个羧酸铑催化剂的构型具有决定性的作用,以下就是典型的很知名的三种构型的手性羧酸铑催化剂(Figure 3)。它们分别具有D2对称(up-down-up-down),C2对称(up-up-down-down),C4对称(all-up)的对称构型。这两大类的手性羧酸铑催化剂分别来自于Emory Univ的Davies与北海道大学的Hashimoto之手,后者已经退休,前者还在继续发光发亮,虽然也一把年纪了,小编在学会都见过两位大神并且聊过,聊完只能是仰望。(在这里小编要补充一下,就prolinate配体而言,Michael.P.Doyle教授的Rh2(MEPY)4催化剂 6与M.A.McKervey教授的rhodium(II) N-benzenesulfonyl-L-prolinate催化剂 7要早于Huw.M.L.Davies教授的Rh2(DOSP)4)
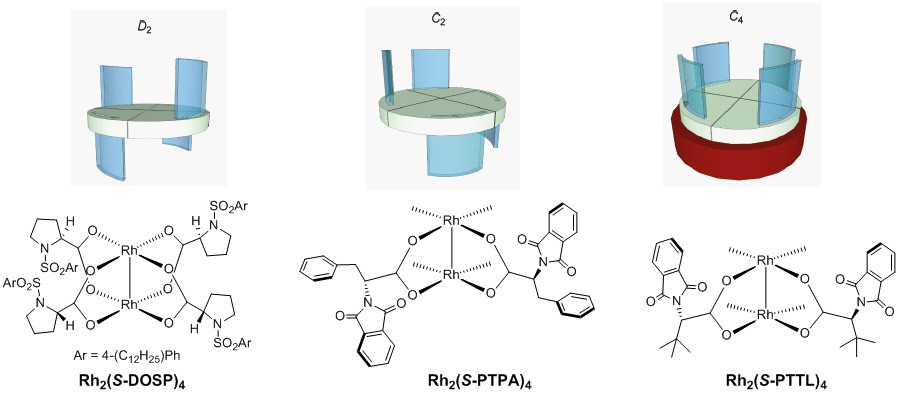
Figure 3. Three distinct ligand orientations used to rationalize enantioselectivity in dirhodium carboxylate-catalyzed reactions.
羧酸铑催化的C-H插入反应中底物的基本反应性问题
从本文一开始的机理介绍中我们可以看出卡宾活性中间体的卡宾C是显略正电性的,所以比较容易与相对富电子的C-H进行C-H插入反应(Scheme 2),因此就电子效应的影响来看,三级C-H活性>二级C-H>一级C-H; 但是另一方面,由于配体等立体位阻的影响,从立体方面考虑活性又是相反的,所以这个balance的问题就得具体底物具体分析了。(重要!)这里的所有底物活性探讨都是基于donor/acceptor类型的重氮化合物(即重氮化合物左右的取代基分别是EWG和EDG)。而如果都是acceptor/acceptor型的重氮底物的话,基本只受电子效应的影响。
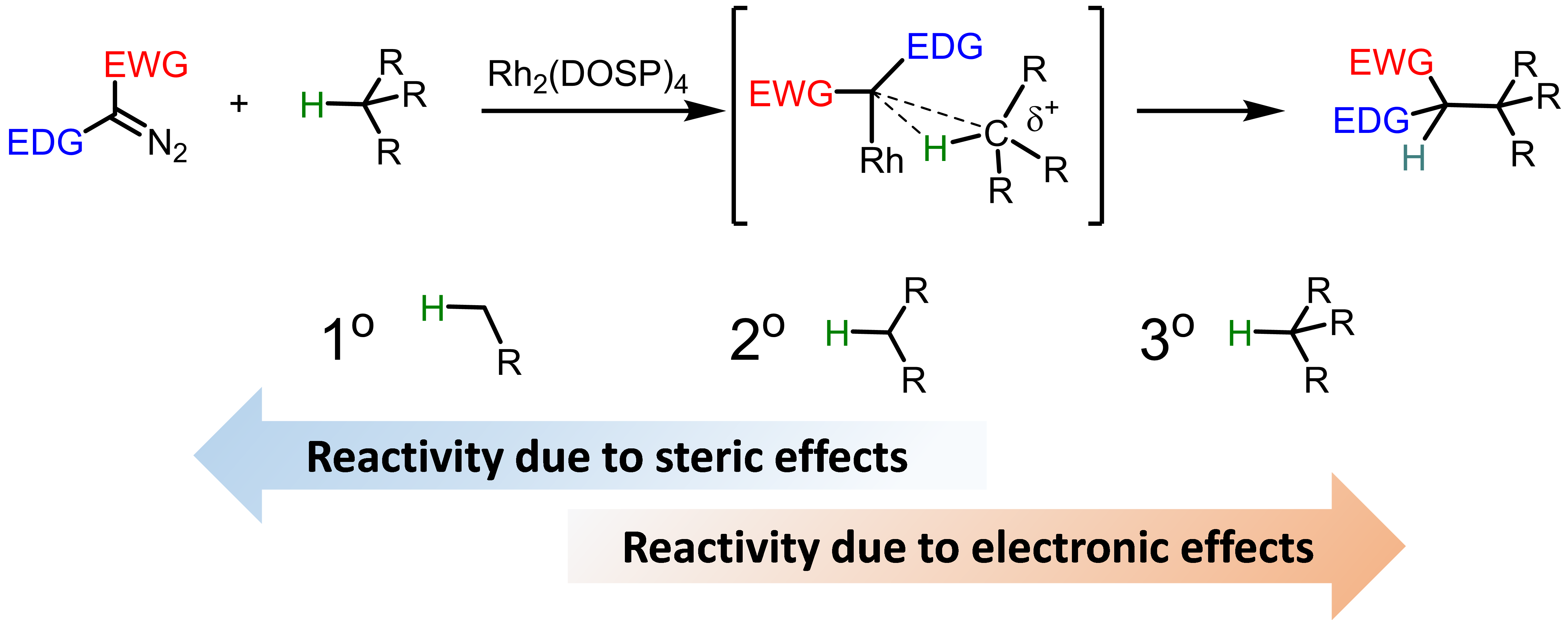
Scheme 2. Controlling elements for site selectivity in donor/acceptor rhodium carbene C–H insertion
以下是一些底物的竞争性C-H活化反应的数据5(Scheme 3),从具体数据更有助于理解一般底物的反应特性,当然再次补充下,这是基于donor/acceptor重氮底物的C-H插入反应。从下图可以看出环己二烯是环己烷的26000倍,比苯乙烯的环丙烷化的活性更高。而杂原子的alpha-H相对于富电子,因此四氢呋喃或者NBoc四氢吡咯的反应活性分别是2700,1700倍,显而易见,对于同为二级C-H的这些底物,富电子的C-H的活性更强。而对于最右侧的2-甲基丁烷或者2,3-二甲基丁烷,虽然三级C-H相对于更富电子,但是由于位阻的问题,反而反应性降低了。从这些数据可以看出,对于donor/acceptor重氮底物来说,收到电子效应与位阻效应双重影响,所以比较难实现site-selective的C-H插入反应。也正因为这样,才使得这种逆自然的反应更加有挑战性。
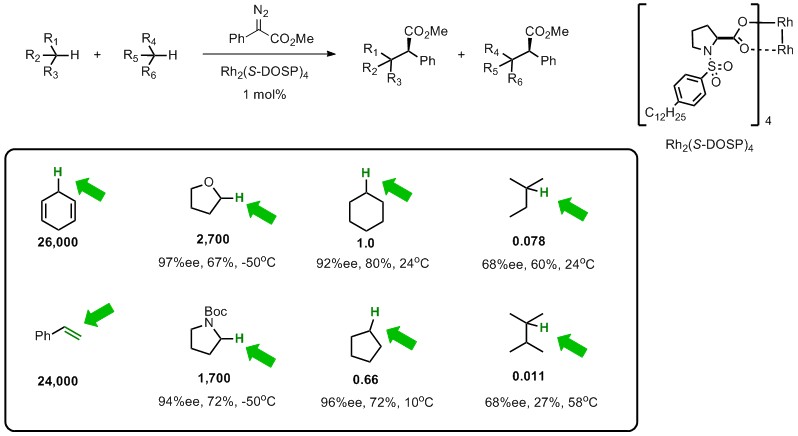
Scheme 3. Relative rates of reactivity for rhodium carbene C–H functionalization.
小编的话
这一篇就简单介绍下羧酸铑催化的卡宾的C-H插入反应的基础,为什么这么长时间小编没有再次写一些深度的论文介绍,实在是小编最近忙着各种实验,学会,攒数据写文章等等,对于一个即将毕业的博士,请大家谅解。
下一篇小编准备具体介绍下最近 Huw M. L. Davies发的那篇Nature 533, 230–234,敬请期待。
参考文献
- Kodadek, T. et al, Science 1992, 256, 1544-1547.
- Weiming Wu. et al, Org. Lett., 2007, 9, 1663.
- Yamanaka, M. et al, J. Am. Chem. Soc. 2002, 124, 7181.
- Alois Furstner. et al, J. Am. Chem. Soc. 2016, 138, 3797.
- Huw M. L. Davies. et al, J. Am. Chem. Soc., 2000, 122, 3069
- M. Anthony McKervey and Tao Ye., J. Chem. Soc. Chem. Commun. 1992, 823.
- M. P. Doyle. et al, J. Am. Chem. Soc., 1991, 113, 8982.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!

















No comments yet.