
作者:石油醚
导读:
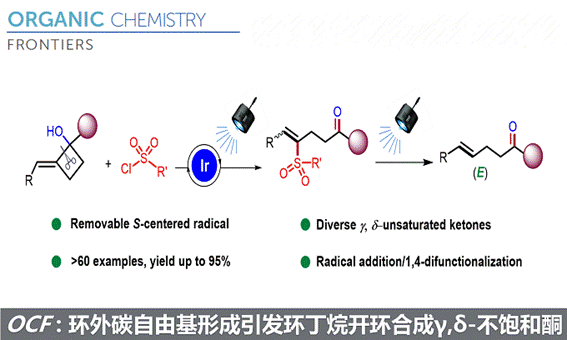
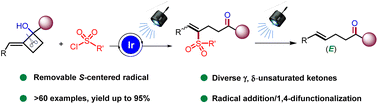
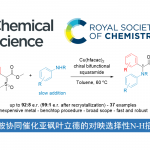
近期,浙江师范大学的张岩课题组在Org. Chem. Front.上发表论文,利用光诱导C-C活化和应变张力释放的策略,实现了环外碳自由基形成引发环丁烷开环合成γ,δ-不饱和酮。
“Photoredox-enabled ring-opening of cyclobutanes via the formation of a carbon radical.
Chunhang Zhao, Wenjing Ma, Kairui Liu, Ruoyang Xu, Xiuya Ma and Yan Zhang *.
Org. Chem. Front., 2024,11, 4663-4670. Doi: DOI: 10.1039/D4QO00996G
研究背景
环丁烷及其取代的环丁烷在结构上是具有较高环张力的骨架,可利用其选择性C–C键活化发生扩环或者开环,用以实现官能团转化或者复杂分子的合成。事实上实现形式上的环丁烷骨架扩环有多种方式,首先可以利用若干人名反应中碳正离子的重排过程,如Demjanov重排、Wagner-Meerwein反应、Baeyer-Villiger 反应以及Pinacol重排。但每一种方法都需使用结构限定的反应物,因此发展出更多有价值的实现环丁烷中惰性C–C键活化的合成策略仍是十分必要的。其中,通过单电子转移引发的自由基反应实现环丁醇开环代表着一类经典的合成策略,其主要特点是环丁醇氧化形成高活性环丁氧自由基并诱发邻位C–C键断裂。但为实现环丁烷开环产生丰富的结构多样性,寻找新模式下的实现环丁烷C–C键活化的自由基反应转化策略仍是极富挑战性的研究课题。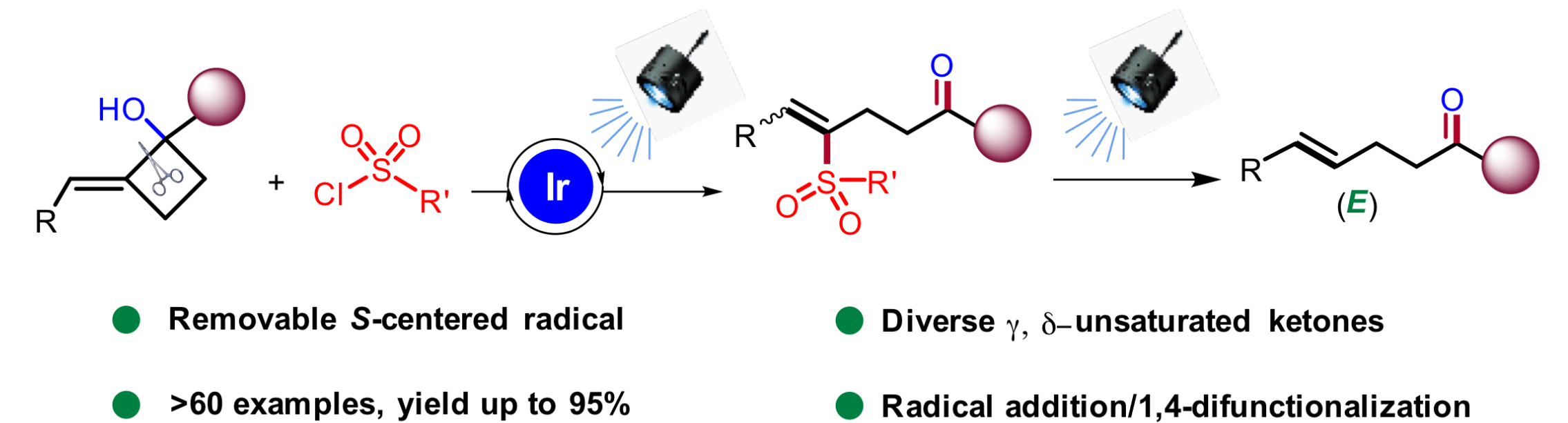
研究内容
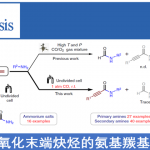
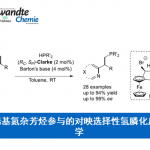
张岩课题组从更普遍的环外碳自由基优先形成出发,思考环丁基亚甲基诱发环丁烷开环的可能性。需要指出的是,与之前的策略相比较,这种开环策略得到的产品中双键是保留的,同时实现远端官能团化。本设计的应用空间理论上会更大,并且能够产生一系列结构新颖的远端双官能团化的烯烃衍生物。基于此研究思路,张岩课题组前期已经完成了底物设计与转化,并实现了γ,δ-不饱和醛类化合物的模板化合成(Angew. Chem. Int. Ed. 2023, 62, e202300166)。严格来说,此次报道的工作是对前期研究发现的很好补充。通过改变反应物结构的调整,成功实现了用传统方法难以合成的γ,δ-不饱和酮类化合物。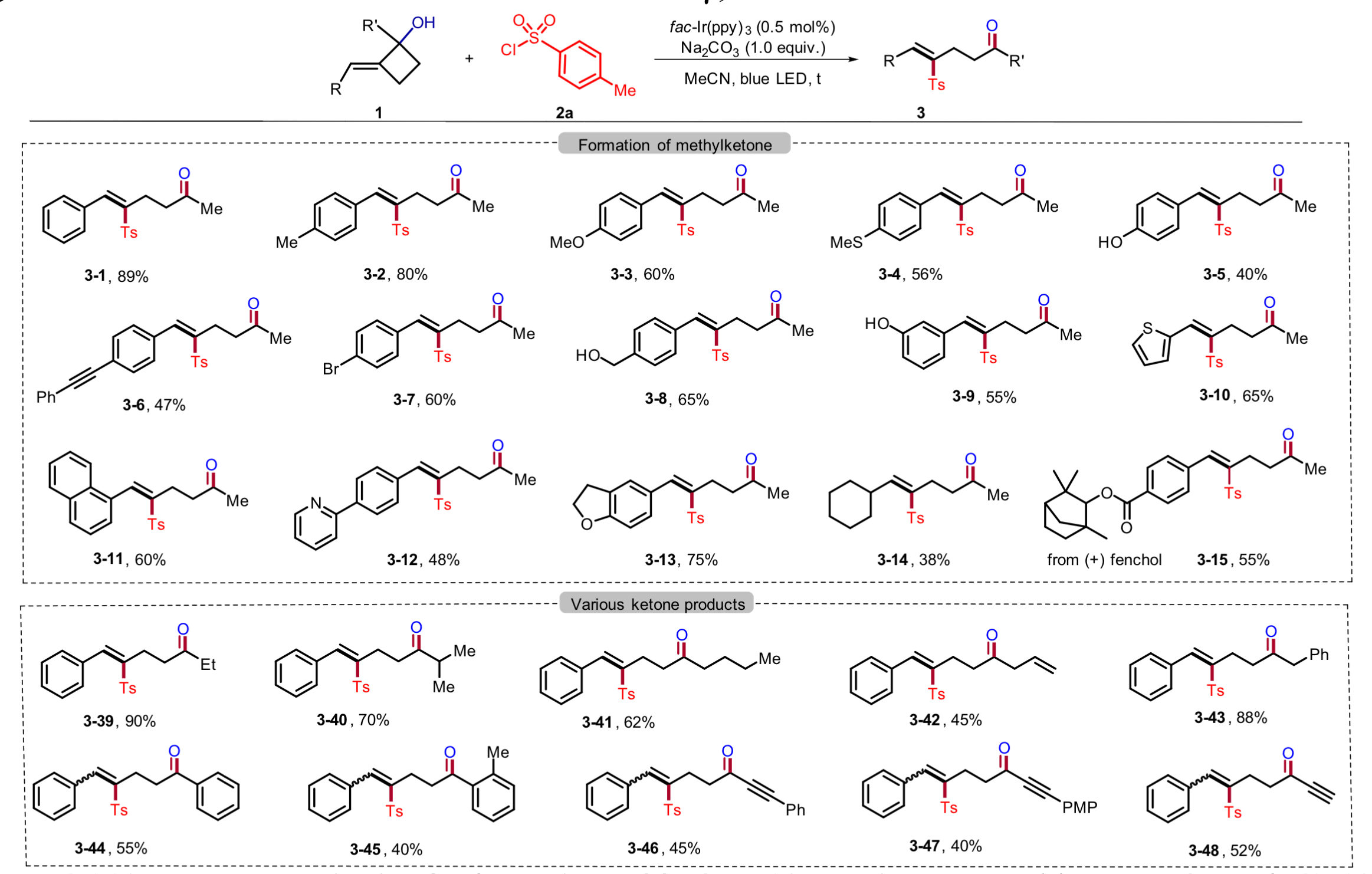 同样的,课题组仍然考察了自由基片段的可移除性。并且无论反应物的烯基片段是Z还是E构型,在光催化脱去自由基片段之后都会变为E型产物。最后,作者还对产品的结构进行了若干衍生化实验,转变为其他有价值的1,4-双官能团产品。
同样的,课题组仍然考察了自由基片段的可移除性。并且无论反应物的烯基片段是Z还是E构型,在光催化脱去自由基片段之后都会变为E型产物。最后,作者还对产品的结构进行了若干衍生化实验,转变为其他有价值的1,4-双官能团产品。 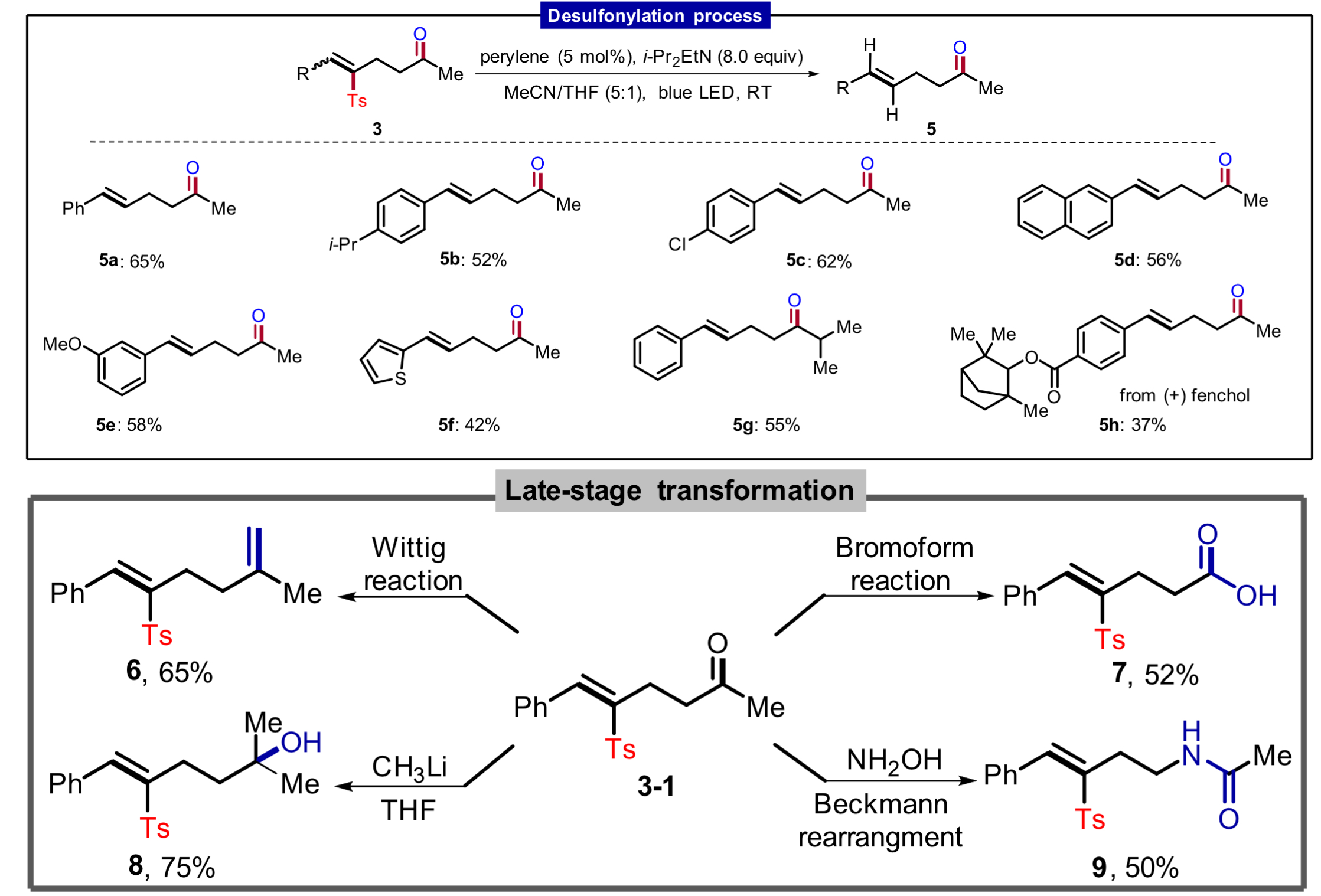
总结展望
总之,本研究工作中促进开环或者说实现环丁烷C–C键活化的因素主要有两个,一是环丁烷骨架的张力驱动;二是形成了由稳定化基团作用的热力学更为稳定的新自由基物种。课题组所建构起来的环丁烷开环的新的转化模式,将为自由基引发的1,4-双官能团化反应的发展提供新的思路和启示。
(非常感谢张岩教授对Chem-Station的支持)
通讯作者简介

张岩,浙江师范大学化材学院,副教授,硕导。2016年毕业于浙江大学药学院,2019年作为国家公派访问学者前往德国哥廷根大学学习。实验室研究方向集中于光或电催化促进的自由基绿色生成方法及其转化。自独立工作以来,以第一作者或者通讯作者在国际顶级及权威期刊发表论文19篇,包括Angew. Chem. Int. Ed.(2篇), Chem. Sci.(3篇), Green Chem.(1篇),Sci. China Chem.(1篇),Org. Lett.(4篇)以及Org. Chem. Front. (2篇)等。授权发明专利12项(含2项美国发明专利)。
















No comments yet.