

本文作者:杉杉
导读
近日,苏黎世大学(University of Zurich)Cristina Nevado教授课题组在Angew. Chem. Int. Ed.上发表论文,报道了镍催化乙烯基酰胺的不对称还原氢芳化(hydroarylation)反应,实现了对映体富集的α-芳基苯甲酰胺化合物的合成。手性双咪唑啉(BIm)配体与二乙氧基甲硅烷、芳基卤化物的结合使用,可将芳基区域选择性地引入烯烃的内部,并在N原子上的α位形成新的立体中心。该反应可在中性且温和的条件下反应,并为抗癌生物活性分子、SARS-CoV PLpro抑制剂和KCNQ通道打开剂的合成提供了简便方法。
Nickel-Catalyzed Asymmetric Synthesis of α-Arylbenzamides
Sergio Cuesta-Galisteo, Johannes Schörgenhumer, Xiaofeng Wei, Estibaliz Merino, and Cristina Nevado
Angew. Chem. Int. Ed. ASAP DOI: 10.1002/anie.202011342

正文
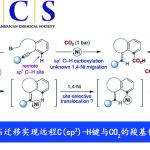
α-芳基苯甲酰胺作为生物活性分子中常见的骨架,如抗癌药、SARS-CoV PLpro抑制剂和抗抑郁药(Scheme 1a)。然而,对于此类骨架的不对称合成则具有难度。目前已有一些课题组成功合成此类结构,如Rh催化乙烯基酰胺加氢反应[1],羧酸衍生物与手性α-芳基取代胺之间的C-N偶联[2],N-酰基氮丙啶氢化物开环[3]等。然而,由于底物范围窄、反应条件苛刻以及差向异构化的问题从而限制了应用。最近,通过镍/光氧化还原双重催化,也实现苯甲酰胺的α-芳基化反应[4](Scheme 1b)。此外,通过三组分(涉及乙烯基苯甲酰胺、卤代芳烃和氢源)的氢芳化反应也可获得上述产物(Scheme 1c)。尽管如此,烯烃的不对称加氢芳化反应仍然研究较少。据报道,通过Pd或Cu配合物也可实现芳基卤化物或硼烷的反应,从而获得此类结构[5]。最近,关于Ni催化高度选择性的分子内环化也被报道[6]。尽管已取得一定的成果,但这些转化大部分仅适用于苯乙烯类底物,因此,将芳基引入杂原子的α位仍是一项难以克服的挑战。近期报道的烯烃不对称双官能化反应[8]中,手性双噁唑啉配体和烯烃上配位点的结合可确保Csp3-Csp2键立体中心的形成。基于上述发现以及该课题组对镍催化π-体系的高效双官能化的持续研究[7],他们开始对官能团化的烯烃(尤其是乙烯基酰胺)进行不对称氢芳化反应的研究。从机理上讲,通过硅烷与镍的活化作用,可以在双键上生成手性Csp3-Ni(I)中间体。此外,通过氧化加成活化Csp2-X前体,形成手性烷基芳基Ni(III)配合物I。C-C键形成的还原性消除发生在烯烃的内部,从而获得具有高立体选择性的产物。因此,需将Csp2-卤化物和氢化物引入到乙烯基苯甲酰胺中使其具有高区域选择性和立体选择性。该过程避免了敏感的有机金属试剂的使用,可在室温下反应,获得对映体富集的α-芳基苯甲酰胺化合物,为后续的衍生化提供了多种可能。
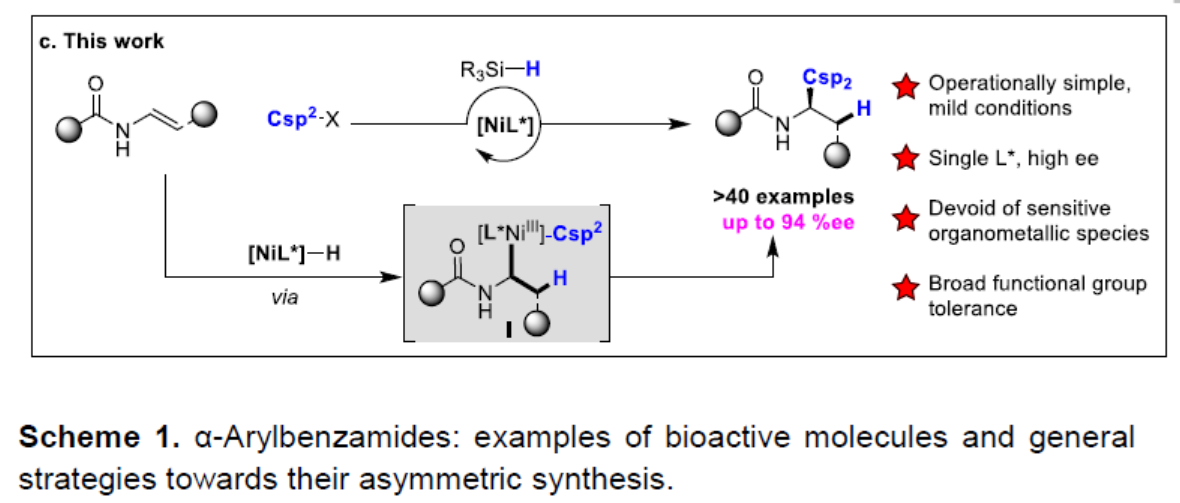
通过相关手性配体的筛选后(Scheme 2),作者发现,在配体L15时,可获得58%收率和94%ee的目标产物1。相同条件下,也可获得72%收率和89%ee的目标产物2。
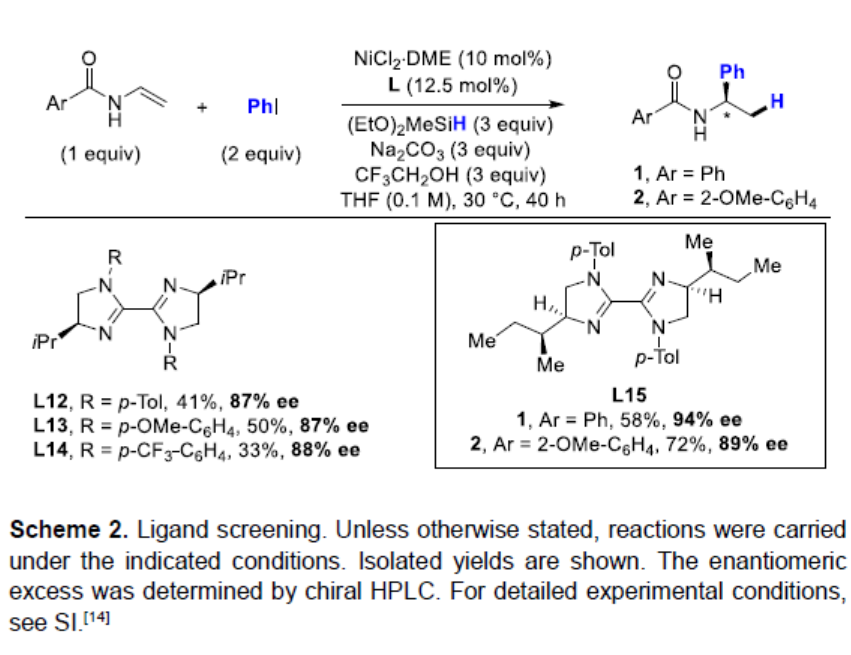
在获得上述最佳反应条件后,作者开始对卤化物进行扩展(Scheme 3)。首先,在对位上具有不同取代的苯基碘化物,如给电子基团(Me,OMe,tBu)和吸电子基团(CN,F,CO2Me,COMe)均可以耐受,获得相应的产物4–10。同样,在间位取代基(甲氧基,3,5-二氟和3-氟和3,5-二甲基二羧酸酯),也可获得产物11–14。同时,一些经典交叉偶联反应中通常使用的官能团,如硼烷、卤素等,均可与体系兼容,从而获得产物15–18,为后期修饰提供了多种可能。此外,延长反应时间和使用4当量芳基碘化物,方可获得产物19–20。值得注意的是,活化的Csp2-溴化物(如2-溴-1H-茚)也可以在标准反应条件下生成所需的产物21,β-溴苯乙烯也可获得相应的酰胺产物22,但立体选择性低。此外,具有立体中心更为复杂的底物也与体系兼容,如L-苯丙氨酸、雌酮和D-葡萄糖衍生的芳基碘化物,均以非对映异构体比率(d.r. ≥ 95:5)获得产物23–25。而在经典的Pd催化交叉偶联反应条件下,化合物18中Csp2-I键衍生化后以76%的产率获得产物26,且对映体纯度未受影响。有趣的是,苄溴也可以用于反应,以90%的收率获得产物27,但ee仅为60%。
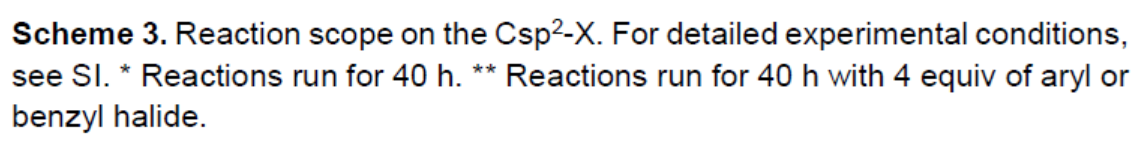
随后,作者对乙烯基酰胺底物进行了相关的扩展(Scheme 4)。未取代(28)、邻甲基(29,30)、邻氯(31,32)、间氯(35,36)、邻氟(33,34)取代的苯甲酰胺均为良好的底物。同时,在对位上具有给电子基团(OMe,Me)和吸电子基团(CF3,Cl)的苯甲酰胺,均可获得相应的产物37–43。此外,含吡啶的底物也可获得所需的手性酰胺44(39%)。而烷基乙烯基酰胺底物,需要延长反应时间,才能获得产物45。值得注意的是,(E)或(Z)-苯甲酰胺底物具有明显不同的反应性,影响产物46的收率,如E型收率仅为29%,而Z型收率为54%,但ee均为94%。同样,(E)-苯乙烯基苯甲酰胺以中等收率获得产物胺47,但是具有优异的对映选择性(96%ee)。

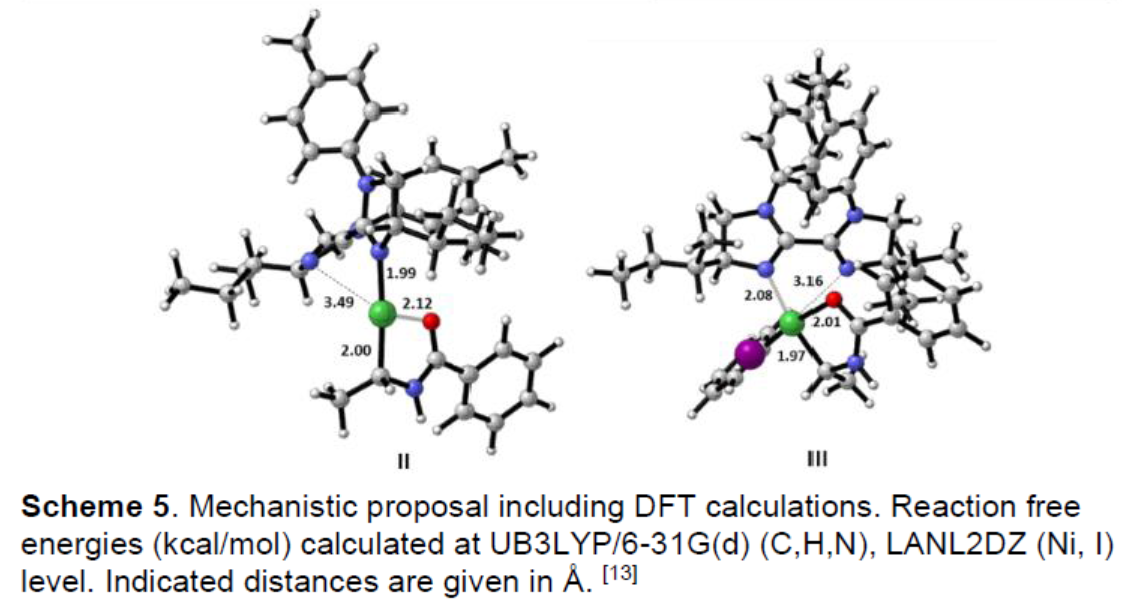
根据对照实验和相关的DFT计算,作者提出了一种可能的反应机理(Scheme 5)。首先,硅烷Me(OEt)2SiH与镍配合物V反应生成氢化镍配合物I。 随后,乙烯基苯甲酰胺插入,形成中间体II。紧接着,经氧化加成和还原消除,即可获得目标产物IV。值得注意的是,酰胺基团对于高立体选择性和区域选择性至关重要,并且烯烃的插入在氧化加成和还原消除之前发生。
总结
苏黎世大学Cristina Nevado教授课题组报道了通过镍催化,实现乙烯基酰胺底物高效不对称还原氢芳化反应,可在室温条件下,获得对映体富集的α-芳基苯甲酰胺化合物。同时,该反应涉及关键的手性双咪唑啉配体(BIm)的烷基Ni(III)中间体。而DFT计算表明,酰胺基在稳定Ni中心中起关键作用,有助于形成立体的Csp3-Csp2键。此外,该方法可有效的合成抗癌生物活性分子、SARS-CoV PLpro抑制剂和KCNQ通道打开剂。

















No comments yet.