
本文作者:杉杉
导读
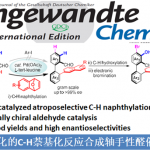
近日,中国科学院广州生物医药与健康研究院朱强课题组在J. Am. Chem. Soc.上发表论文,报道了一种钯催化N-烷基-2-异氰基苯甲酰胺与2,6-二取代芳基碘化物的环酰胺化反应,从而获得轴手性2-芳基喹唑啉酮衍生物,具有良好的收率和阻转选择性。同时,当以N-(2,4-二甲氧基苯基)-2-异氰基苯甲酰胺作底物时,会产生具有两个立体异构轴的2,3-二芳基喹唑啉酮,具有中等的非对映选择性和良好的对映选择性。其中,中国科学院广州生物医药与健康研究院朱强和罗爽为共同通讯作者。
Palladium-Catalyzed Atroposelective Coupling-Cyclization of 2‑Isocyanobenzamides to Construct Axially Chiral 2‑Aryl- and 2,3-Diarylquinazolinones
Fan Teng, Ting Yu, Yan Peng, Weiming Hu, Huaanzi Hu, Yimiao He, Shuang Luo,* and Qiang Zhu*
J.Am. Chem. Soc.ASAPDOI:10.1021/jacs.1c00640

正文
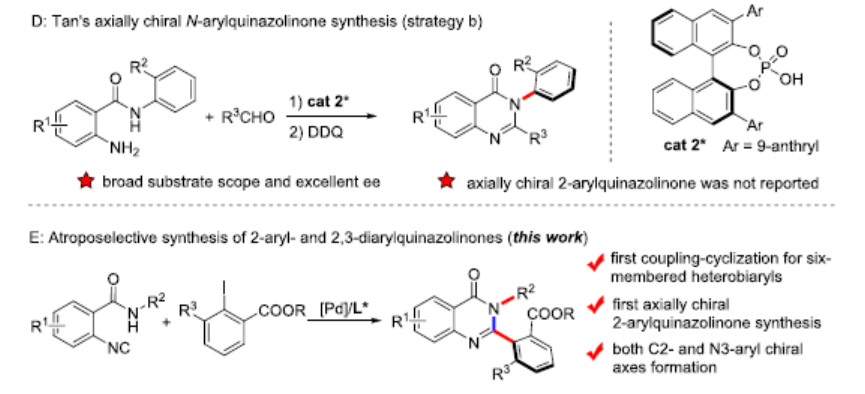
轴手性联芳基化合物广泛存在于手性配体或催化剂以及生物活性分子中,目前已开发出多种催化方法来构建联芳基骨架,主要基于以下三种策略:(a)两个现有芳基的对映选择性偶联;(b)通过立体选择性环化或通过点到轴的手性转移从而构建芳环;(c)通过不对称去对称化、C-H官能化、开环等修饰前手性联芳基以形成手性联芳基。然而,对于涉及偶联-环化策略却很少被研究(Scheme 1A)。2019年,Li等[1]报道了一种通过手性Rh(III)配合物催化原位形成吲哚后再进行C-H芳基化从而实现轴手性联吲哚的合成(Scheme 1B)。同年,Kato等[2]通过Pd(II)催化邻炔基苯硫醚的环化和二聚化从而构建阻转异构联苯并噻吩化合物(Scheme 1C)。然而,除了五元联杂芳基外,该策略在其它轴手性联芳基合成中的应用仍在探索中。
喹唑啉酮衍生物广泛存在于天然产物和生物活性分子中,具有抗癌、抗炎、抗抑郁等特性。2015年,Miller等[3]报道了一种新型肽催化NBS促进富电子3-(3-羟苯基)-2-烷基喹唑啉酮的溴化反应,以获得具有高ee的高度阻转异构稳定性的三溴化3-芳基喹唑啉酮。后来,Kitagawa等[4]报道了Pd催化的3-(2,6-二溴苯基)-喹唑啉酮的不对称去对称脱溴化反应,从而获得单溴化3-芳基喹唑啉酮。2017年,Tan等[5] 报道了一种通用且实用的方法,可通过CPA催化氨基酰胺与醛环化反应合成轴手性3-芳基喹唑啉酮(具有优异的ee和广泛的底物范围)(Scheme 1D)。然而,此类反应主要集中2-芳基喹唑啉酮中轴手性N-芳基骨架的构建,而对轴手性C-芳基骨架的构建却很少。在此,本文报道了一种钯催化N-烷基-2-异氰基苯甲酰胺与2,6-二取代芳基碘化物的偶联-环化反应,可获得轴手性C-芳基喹唑啉酮衍生物,具有良好的收率和阻转选择性(Scheme 1E)。
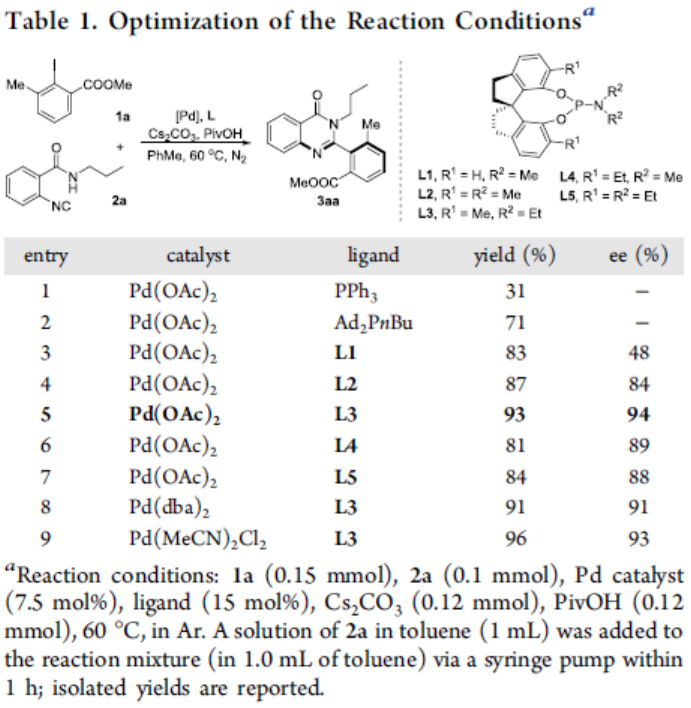
首先,作者以2-碘-3-甲基苯甲酸甲酯(1a)和2-异氰基-N-丙基苯甲酰胺(2a)作为模型底物,进行了相关反应条件的筛选(Table 1)。反应的最佳条件为,以Pd(OAc)2为催化剂, L3为配体, Cs2CO3 为碱,可在60 °C下反应,获得93%收率和94%ee的产物3aa。
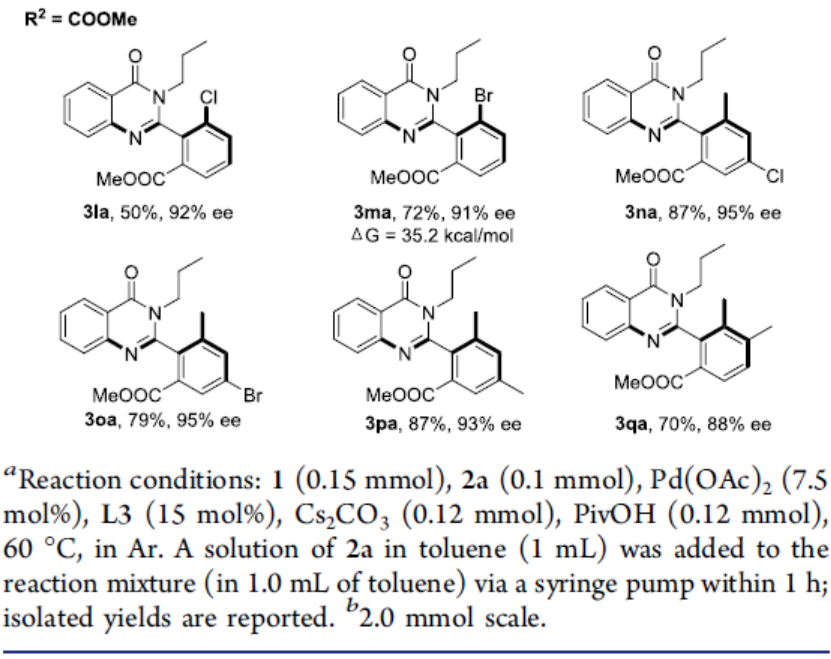
在获得上述最佳反应条件后,作者开始对芳基碘化物底物1进行了扩展(Scheme 2)。当R1为甲基时,酯基R2取代可从甲基逐渐增加到叔丁基,获得相应的产物3aa–3fa,ee不受影响,但收率急剧下降。当R2取代为苯基、苄基和三氟乙基酯时,也可顺利反应,获得产物3ga–3ia。对于R2取代为N,N-二甲基酰胺(3ja)或N-甲氧基-N-甲基酰胺(3ka)时,ee值均有所下降。对于R2取代为乙酰基时,未能进行反应。此外,当R2取代即为甲酸甲酯时,芳香环上含有Cl、Br和Me时,均可顺利反应,获得相应的产物3la–3qa。
随后,作者对异氰化物底物2进行了相关的扩展(Scheme 3)。当R2取代为乙基、丁基、环戊基、苄基时,可获得产物3ab–3ae。同时,底物中的芳环不受电子效应和定位效应的影响,均可获得良好收率和ee的相应产物3af–3aj和3am。
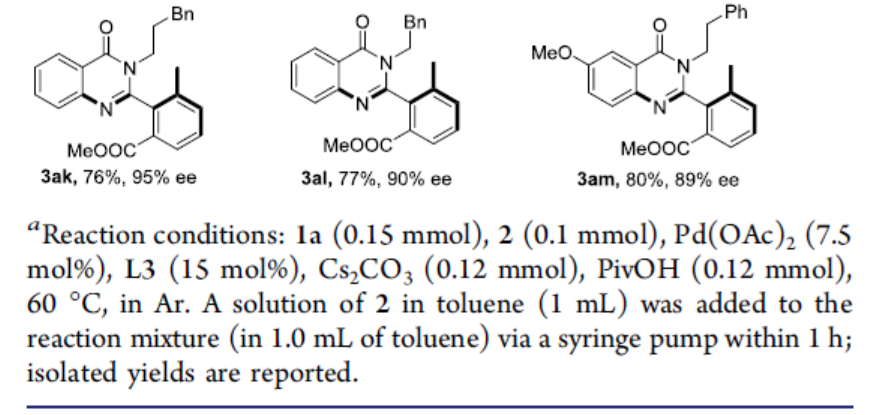
同时,通过上述方案可实现具有两个立体轴喹唑啉酮衍生物的构建(Scheme 4)。因此,作者以4和1a为底物,进行了相关的反应,从而获得两种所需的非对映异构体5a(收率70%,ee为93%)和5b(收率21%,ee为89%)。

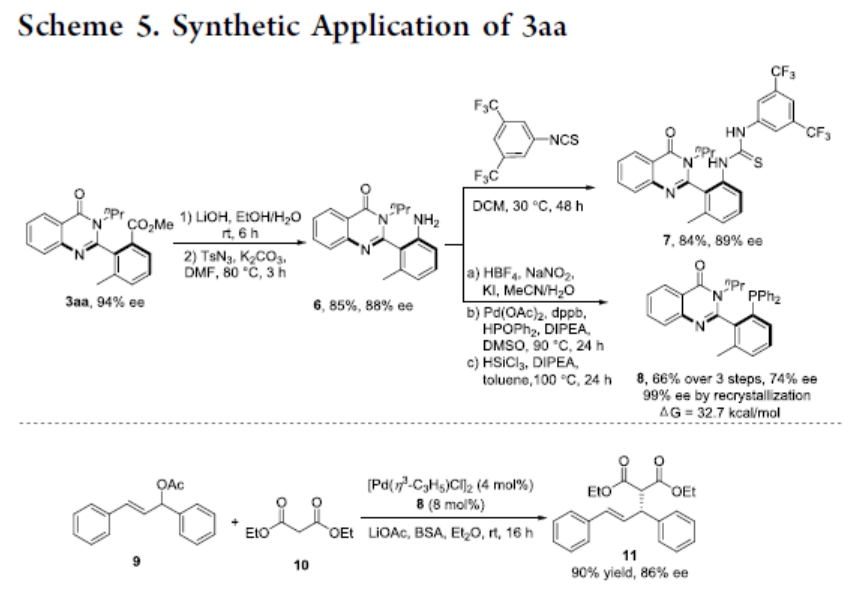
随后,作者对反应的实用性进行了研究(Scheme 5)。首先,3aa经水解和Curtius重排可获得85%收率的苯胺6。6可与1-异硫氰基-3,5-双(三氟甲基)苯经缩合反应形成富含阻转异构的硫脲7。6通过Sandmeyer碘化、Pd催化的C-P键的形成和还原反应,可合成化合物8,尽管在三步中观察到手性的损失,但经重结晶后可获得对映异构纯的膦8。此外,手性膦8可用于Pd催化9和10的不对称烯丙基烷基化反应,以90%的收率和86%ee提供所需的产物11。
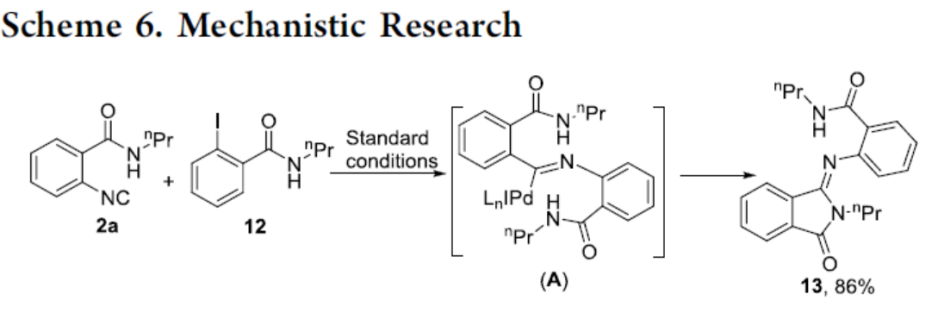
为了进一步了解反应的机理,作者以2-碘-N-丙基苯甲酰胺(12)和2a在标准条件下反应,经中间体A,获得86%收率的形成异吲哚啉酮衍生物13,从而表明在氧化加成后将异氰酸酯插入Pd(II)中间体是一种有利的方法。因此,反应很可能通过偶联-环化而不是环化-偶联的顺序进行(Scheme 6)。
总结
中国科学院广州生物医药与健康研究院朱强课题组报道了一种钯催化N-烷基-2-异氰基苯甲酰胺与2,6-二取代芳基碘化物的环酰胺化反应,可获得轴手性2-芳基喹唑啉酮衍生物,具有良好的收率和阻转选择性。同时,当以N-(2,4-二甲氧基苯基)-2-异氰基苯甲酰胺作底物时,会产生具有两个立体异构轴的2,3-二芳基喹唑啉酮,具有中等的非对映选择性和良好的对映选择性。此外,反应涉及偶联-环化的过程。


















No comments yet.