

本文作者:石油醚
导读
在化学领域中,反应过程动力学分析(RPKA)是众多动力学技术中的一个子集,用于确定化学反应的速率规律与阐明化学反应的机理。虽然指导反应过程动力学分析的相关概念并不新鲜,然而,这一动力学分析技术直至在20世纪90年代末才由D. G. Blackmond教授(目前在Scripps研究所工作)正式提出。,并在此后获得越来越广泛的应用1。与更常见的拟一级动力学分析(Pseudo-first-order analysis)不同, 由于反应机理可能因所涉及物种的相对与绝对浓度而发生改变,因此,这种方法获得的结果比传统方法更能代表相关反应物在通常实验条件下的反应行为。此外,通过观察反应随时间的变化而获得的相关信息,能够进一步发现反应过程中中存在的诱导期(induction periods)、催化剂失活(catalyst deactivation)或机理突变 (changes in mechanism) 等异常行为。
简介
反应过程动力学分析 (RPKA) 可以在反应体系中存在两种或多种反应物的浓度同步改变的合成实验条件下,通过准确原位监测整体反应过程,从而获得大量数据,进而简化动力学研究,事实上,这与实际合成实验过程中,反应参数发生改变的方式一致。RPKA 方法的主要优势便是可以在新反应的早期研究中,快速获取重要的动力学信息,从而有利于指导后续的反应优化与把握反应机理研究的方向1。
监测反应进度
反应过程动力学分析是取决于精确监测反应转化率随时间变化的能力。要实现这一目标,则需要一系列监测技术(最为常见的监测技术接下来小编将作详细介绍)。有时,化学家这些技术分为微分监测技术(监测反应速率随时间的变化)或积分监测技术(监测底物与/或产物含量随时间的变化),并且,通过简单的数学变换(微分或积分),能够将通过两种方法中的任意一种所获得的数据进行相互转换。事实上,不管采用哪一种监测技术,均能够通过选择其他不同的动力学分析方法进一步证实上述监测技术在反应参数分析中的有效性1。
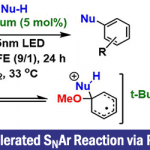
1.反应进程NMR技术
核磁共振波谱通常是监测相关反应过程的首选方法,其中底物的消耗或产物的形成过程均能够通过相对于非反应活性标准物的峰面积积分随时间变化观察到。根据浓度数据,通过采用多项式导数对实验曲线拟合,进而获得反应速率随时间的变化图像2。由于收集的原始数据与浓度以及时间呈正比,因此,这种反应过程的NMR监测技术可以归类为积分技术1(图1 )。尽管这一技术能够清晰方便的分析具有具有特征的、易于分辨的产物或反应物吸收峰的特定反应体系,然而,这一技术的弊端是需要一个能够在NMR管中反应的均相反应体系。尽管NMR观察可以识别反应中间体,而且,反应过程中任何给定物种的存在并不一定表明其参与相应反应过程。然而,反应进程NMR通常在变温条件下进行,从而能够将反应速率调节至便于观察的水平。目前为止,已有较多采用反应进程NMR研究化学反应的实例。最具代表性是实例为Buchwald–Hartwig胺化反应的研究3。
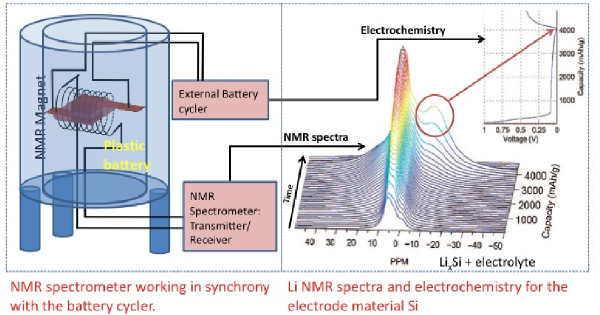
图1 反应进程NMR
2.原位红外技术
原位红外光谱法(in situ FT-IR)可用于监测反应过程,前提是试剂或产品在红外光谱区域存在特征的IR吸收。反应物消耗速率或产物生成速率可以通过吸光度随反应时间的改变而获取(应用Beer定律)。即使反应物与产物的吸收光谱显示出一定程度的重叠,只要所研究的IR吸收峰的绝对吸光度随时间能够发生显著变化,现代仪器芳香软件通常可以准确地对重叠部分的相对贡献进行去卷积(deconvolution)分析2。由于收集的原始数据与浓度以及时间成正比,因此,原位红外光谱属于积分监测技术1。根据上述数据,可以通过简单地对实验曲线进行多项式积分拟合的方式获得原料或产物浓度随时间变化的信息。随着原位红外光谱仪监控可靠性的增加,近年来,FT-IR的应用越来越广泛。具有代表性的实例主要涉及氨基硫脲催化的非天然氨基酸的不对称Strecker合成与Lewis碱催化的卤内酯化(halolactonization)与环醚化(cycloetherification) 的机理研究4,5。(图2)

图2 原位红外光谱
3.原位紫外-可见光谱
与原位红外实验类似,只要试剂或产物在紫外-可见区域显示出特征的吸收峰,就能够选用原位紫外-可见吸收光谱法(in situ UV-visible absorbance spectroscopy )监测反应进程。由于反应物消耗速率或产物生成速率可根据吸光度随时间的改变而获取(应用Beer定律),因此,原位紫外-可见光谱同样属于积分监测技术。根据所选择的光谱吸收区域,原位紫外-可见光谱技术在无机或金属有机反应体系中的应用要比在纯有机反应中的更为广泛,典型实例主要涉及钐试剂促进的 Barbier 反应6。(图3)

图3 原位紫外-可见光谱
4.反应量热法
反应量热法(Calorimetry)同样可以用于监测反应过程,因为该仪器检测出的反应瞬时热通量(instantaneous heat flux)与反应过程的焓变化直接相关。由于所收集的原始数据与速率与时间成正比,因此,反应量热法属于微分监测技术7,8。根据所收集的原始数据,可以对实验曲线的多项式积分进行拟合,进而获得起始原料或产物浓度随时间变化的信息9。虽然反应量热法相比其他方法应用较少,然而,该方法却是催化剂筛选的有效工具之一10。此外,反应量热法同样能够应用于单个反应的机理研究,典型实例涉及脯氨酸催化的醛类化合物的α-胺化以及钯催化的Buchwald-Hartwig胺化反应3,11。(图4 )
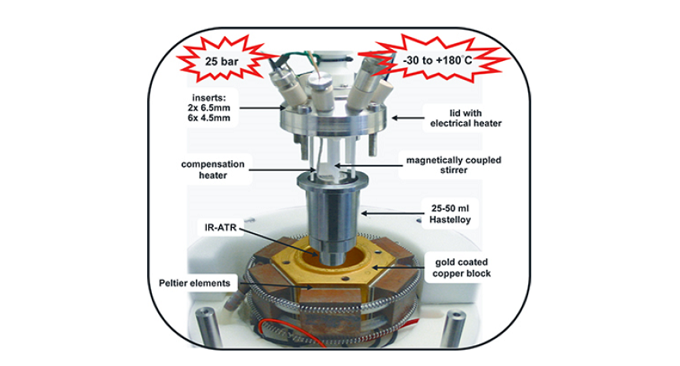
图4 量热技术
5.其他技术
虽然GC、HPLC以及MS法都是分辨一系列不同化合物形成的混合物(有时甚至是对映体混合物)的优良技术,然而,这类测量的时间分辨率不如上述监测技术更加精确。尽管如此,这类监测技术仍然可以用于反应监控,例如在Heck反应的研究中12,反应的非均相特性妨碍了上述监测技术的应用,此外,反应量热法同样能够应用于有机催化剂的SOMO活化的研究13。尽管存在一些不足之处,然而,反应量热技术仍然能够用作之前监控方法一种有力的校准方法。
综上,Chem-Station小编介绍了一系列新的反应监控技术在反应过程动力学分析方面的应用,上述监控技术为RPKA分析提供了强大数据支撑,通过对所得数据的分析,有助于对前期反应的优化以及对反应机理的研究。下期,Chem-Station小编将对反应过程动力学分析的相关原理进行详细描述。
参考文献
- Blackmond, D. G., Angew. Chem. Int. Ed. 2005,44(28), 4302-4320, doi:10.1002/anie.200462544.
- Blackmond, D. G.; Ropic, M.; Stefinovic, M., Org. Process Res. Dev. 2006,10(3), 457-463, doi:10.1021/op060033k.
- Shekhar, S.; Ryberg, P.; Hartwig, J. F.; Mathew, J. S.; Blackmond, D. G.; Strieter, E. R.; Buchwald, S. L., J. Am. Chem. Soc. 2006,128(11), 3584-3591, doi:10.1021/ja045533c.
- Zuend, S. J.; Jacobsen, E. N., J. Am. Chem. Soc. 2009,131(42), 15358-15374, doi:10.1021/ja9058958.
- Denmark, S. E.; Burk, M. T.,PNAS.2010,107(48), 20655-20660, doi:10.1073/pnas.1005296107
- Choquette, K. A.; Sadasivam, D. V.; Flowers, R. A., J. Am. Chem. Soc. 2011,133(27), 10655-10661, doi:10.1021/ja204287n.
- Mathew, J. S.; Klussmann, M.; Iwamura, H.; Valera, F.; Futran, A.; Emanuelsson, E. A. C.; Blackmond, D. G., J. Org. Chem. 2006,71(13), 4711-4722, doi:10.1021/jo052409i.
- Steel, C.; Naqvi, K. R., J.Phys.Chem. 1991,95 (26), 10713-10718, doi:10.1021/j100179a037.
- Blackmond, D. G.; Rosner, T.; Pfaltz, A., Org. Process Res. Dev. 1999,3(4), 275-280, doi:10.1021/op990024u.
- Hein, J. E.; Armstrong, A.; Blackmond, D. G., Org. Lett. 2011,13(16), 4300-4303, doi:10.1021/ol201639z.
- Singh, U. K.; Strieter, E. R.; Blackmond, D. G.; Buchwald, S. L., J. Am. Chem. Soc. 2002,124(47), 14104-14114, doi:10.1021/ja026885r.
- Herrmann, W. A.; Brossmer, C.; Reisinger, C.-P.; Riermeier, T. H.; Öfele, K.; Beller, M., Chem. Eur. J. 1997,3(8), 1357-1364, doi:10.1002/chem.19970030823.
- Devery Iii, J. J.; Conrad, J. C.; MacMillan, D. W. C.; Flowers Ii, R. A., Angew. Chem. Int. Ed. 2010,49(35), 6106-6110, doi:10.1002/anie.201001673.



















No comments yet.