
本文作者:ChemBoy
导读
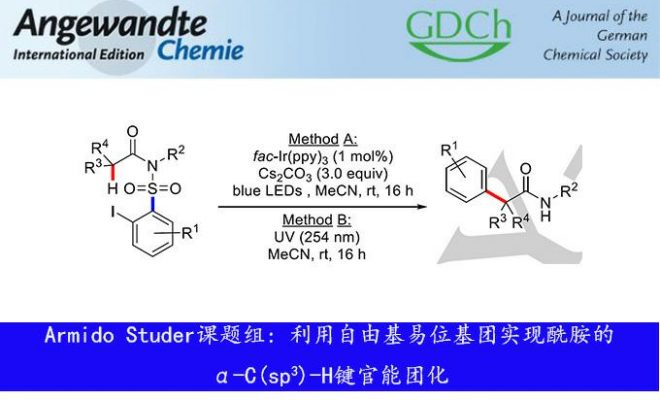
最近,德国明斯特大学Armido Studer教授课题组利用自由基易位芳基化(RTA)基团2-碘芳基砜基在可见光催化条件下成功实现了N-烷基酰胺的α-C(sp3)-H键的芳基化反应。利用该反应可以实现酰胺α-季碳中心的构建。在该反应中,各种单、双取代的RTA基团均能很好地适用于一级、二级和三级α-C(sp3)-H键的芳基化。该自由基转化反应过程中涉及1,6-氢原子转移、1,4-芳基迁移,并且伴随着SO2气体的释放。相关研究成果发表在《Angew. Chem. Ed. In.》上:
“Functionalization of α-C(sp3)‒H Bonds in Amides Using Radical Translocating Arylating Groups”
Niklas Radhoff and Armido Studer*
Angew. Chem. Ed. In., Accepted Article, 10.1002/anie.202013275

正文
前言
过去的三十年里,,C-H键的直接官能团化反应在有机合成化学领域受到了极大的关注。碳氢键的直接官能团化反应是一种具有很高价值的策略,利用过渡金属作为媒介或催化剂进行的C(sp2)-H键直接官能团化反应已经被广泛研究[1]。然而,关于过渡金属催化的远程C(sp3)-H键芳基化反应的报道相对较少[2]。作为过渡金属介导的C-H键官能团化过程的补充,经选择性的氢原子转移(HAT)过程进行的远程自由基C-H建官能团化反应被建立起来[3]。除了反应活性的氮中心自由基可以实现氢原子转移过程,反应活性的碳中心自由基也能应用于氢原子转移介导的远程C(sp3)-H键官能团化反应。早在上世纪八十年代后期,Curran团队在醇和胺类分子上“安装”芳基自由基前体,通过生成的瞬态芳基自由基引发选择性的1,5–HAT过程,生成相应可被捕获的易位的碳中心自由基,从而实现远程C-H键的官能团化[4]。近年来,经过1,n-HAT过程进行的自由基芳基迁移反应开始成为了区域选择性C(sp3)-H键芳基化反应的一种有力工具[5]。在之前,Studer课题组已经发展了利用2-碘芳基砜氯化物作为自由基易位芳基 (RTA) 试剂实现醇类底物的远程自由基C(sp3)-H键芳基化反应(Scheme 1a)[6]。磺酸酯先生成相应的芳基自由基,然后依次发生选择性的1,7-HAT、1,5-芳基迁移和脱砜化,以中等至良好的产率实现醇的γ-C(sp3)-H键芳基化反应。
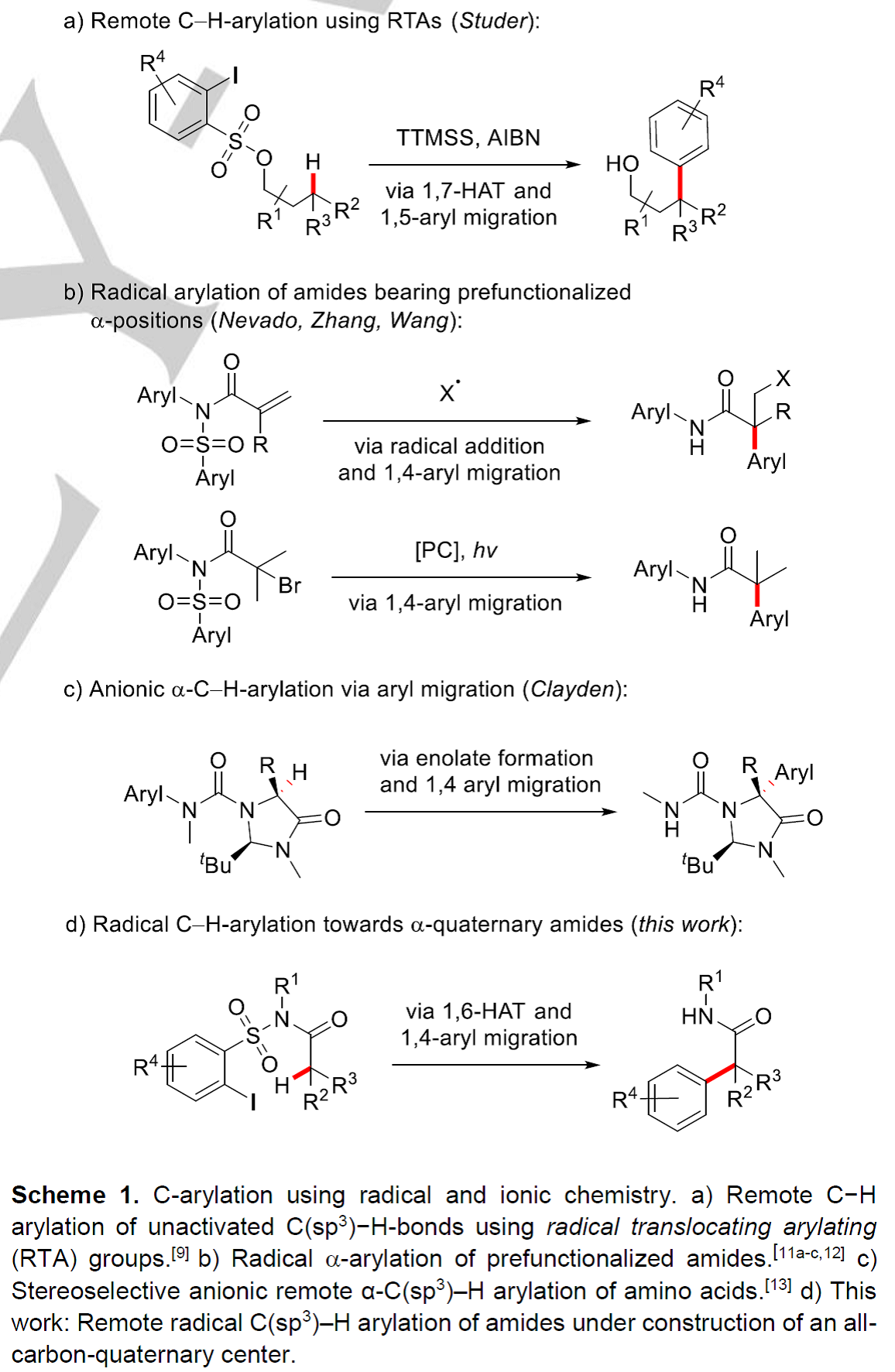
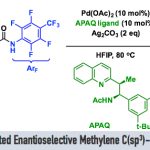
全α-季碳酰胺具有一定的生物活性,因此在药物化学中是一类具有价值的化合物。最近有三种不同的利用自由基化学或离子化学经芳基迁移过程进行酰胺α-芳基化反应的策略被报道。Zhang和Wang课题组发展了一种类似的策略:α-卤代酰胺在光氧化还原条件下被还原生成相应的α-酰胺自由基,然后发生芳基迁移反应(Scheme 1b)[7]。值得注意的是,这两种策略均局限于预功能化的酰胺底物。在2018年,Clayden课题组利用阴离子型的1,4-芳基迁移策略实现了氨基酸类衍生物的立体选择性α-C(sp3)-H键芳基化反应(Scheme 1c)[8]。基于以上这些报道,作者想利用基于N-烷基-邻碘芳基磺酰胺的RTA化学,实现对酰胺类底物进行自由基的α-C(sp3)-H键直接芳基化反应。RTA-基团通过芳基磺酰胺与含α-C(sp3)-H键的酰氯反应就能引入,新生成的磺酰胺通过串联的芳基自由基生成、1,6-HAT、1,4-芳基迁移和还原性的脱砜化就能转化成α-芳基化的酰胺。然而,利用过渡金属催化的策略,一级、二级和三级C(sp3)-H键的直接芳基化反应均具有很大的挑战。
条件筛选
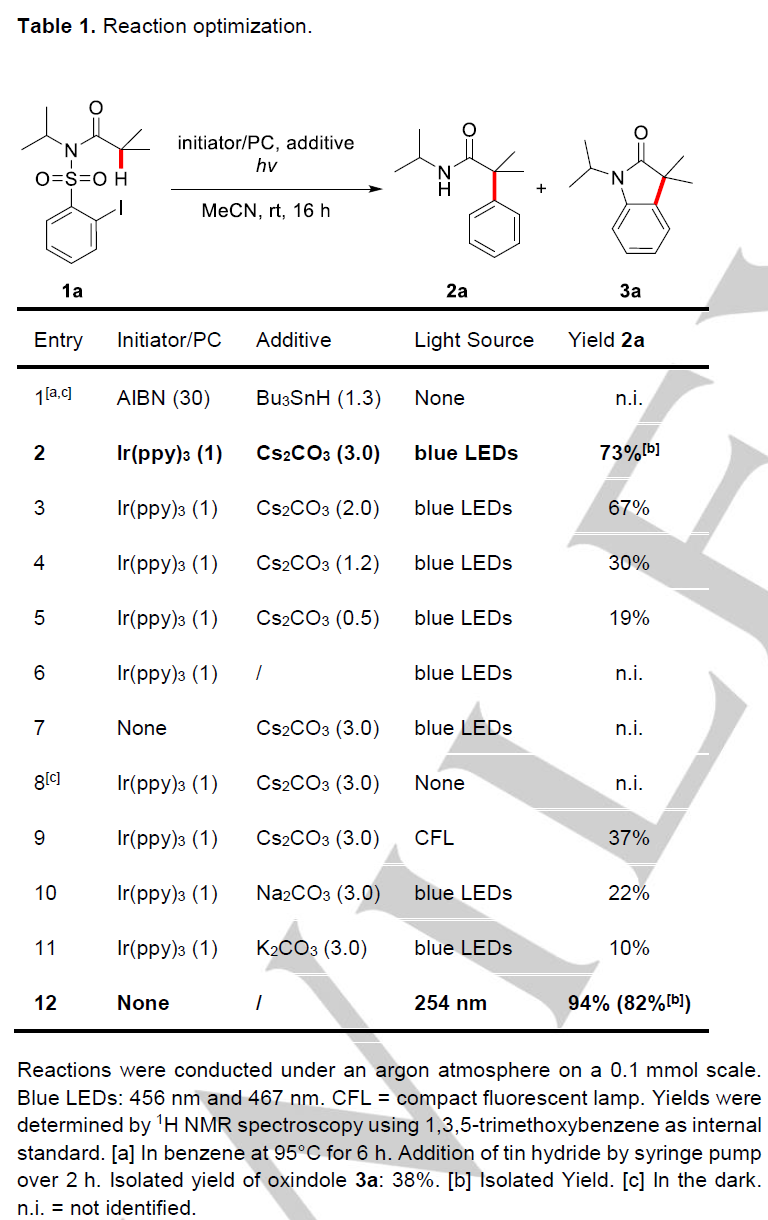
作者以N-2-碘苯磺酰基-N-异丙基异丁酰胺(1a)作为模板底物进行反应条件优化(Table 1)。作者发现,当以偶氮异丁腈(AIBN,0.3eq.)作为自由基引发剂、Bu2SnH(1.2 eq.)作为添加剂、甲苯作为溶剂,95℃条件下回流6h后,未检测到目标产物2a的生成,取而代之,生成的是吲哚酮3a(38%)(Table 1, entry 1)。当作者以Ir(ppy)3(1 mol%)作为光敏剂、Cs2CO3(3.0 eq.)作为添加剂、乙腈为反应溶剂,在蓝光(456 nm and 467nm)辐射下,室温反应16h,能以73%的产率生成目标产物2a, 而无副产物吲哚酮3a生成(Table 1, entry 2, Method A)。随后,作者发现产率会随着Cs2CO3的量减少而降低,当无Cs2CO3时,未检测到目标产物的生成(Table 1, entries 3-6);值得注意的是,在无光催化剂或者是无光照条件下也未检测到目标产物(Table 1, entries 7-8);有意思的是,当在节能灯(CFL)辐射下,该反应也能以37%的产率生成目标化合物2a(Table 1, entry 9);接着,作者分别用Na2CO3(3.0 eq.)和K2CO3(3.0 eq.)代替Cs2CO3(3.0 eq.),发现产率会明显降低(Table 1, entries 10-11)。作者还发现,无光催化剂和添加剂存在时,在254 nm波长的紫外光辐射条件下也能以82%的收率生成目标产物2a(Table 1, entry 12, Method B)。作者以Method A和B两种反应条件作为备选条件对反应的底物适用性进行研究。
底物拓展
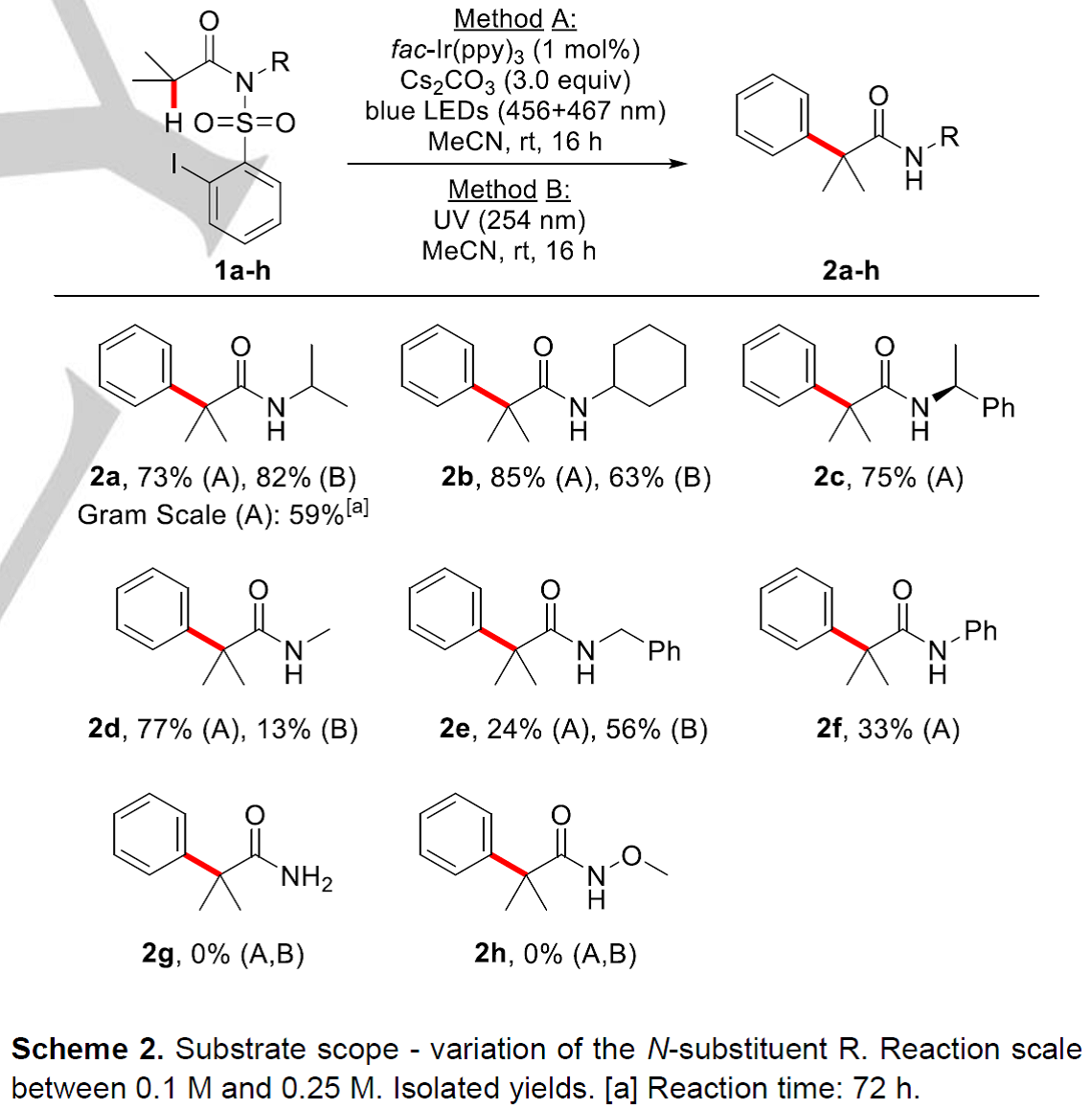
首先,作者就底物的N-R不同取代基对反应的影响进行了研究(Scheme 2)。N-二级烷基取代的磺酰胺类底物1a-c在Method A反应条件下均能以良好的收率获得相应的目标产物2a-c(73-85%),1a-b在Method B条件下也能分别以82%和63%的收率获得2a-b;同时,作者还对1a在方法A的条件下进行了克级反应,能以59%的收率获得2a; 位阻较小的N-Me酰胺在方法A条件下能以77%的收率获得产物2d,而在方法B条件下产率仅为13%;相比之下,N-Bn取代的底物1e在方法A条件下仅有24%的收率,而在方法B条件下有56%的收率;N-Ph取代的底物1f在方法A条件下仅能以24%的收率获得目标产物2f, 而在方法B条件下未检测到目标产物。通过以上结果,作者认为N-取代基的构象效应在该反应中发挥着关键作用。N-原子上无取代的磺酰胺底物1g和Weinreb型酰胺底物1h在方法A和方法B作用下均未检测到相应的目标产物。
其次,作者在方法A的作用下对芳基部分的适用性进行了探索(Scheme 3)。作者发现,苯环上无论是给电子基取代,还是给电子基取代均能以中等至良好的收率获得目标α-C(sp3)-H键芳基化产物(2i-2p, 2s; 48-79%);除了苯磺酰类底物,萘磺酰胺类底物也能顺利进行反应(2q, 73%; 2r, 68%);然而,杂环类底物在该反应条件下不适用(2t and 2u);二酰亚胺取代类底物1v在该反应条件下未生成目标产物,而是经1,6-HAT过程后发生芳香取代反应以76%的收率生成产物2’v。
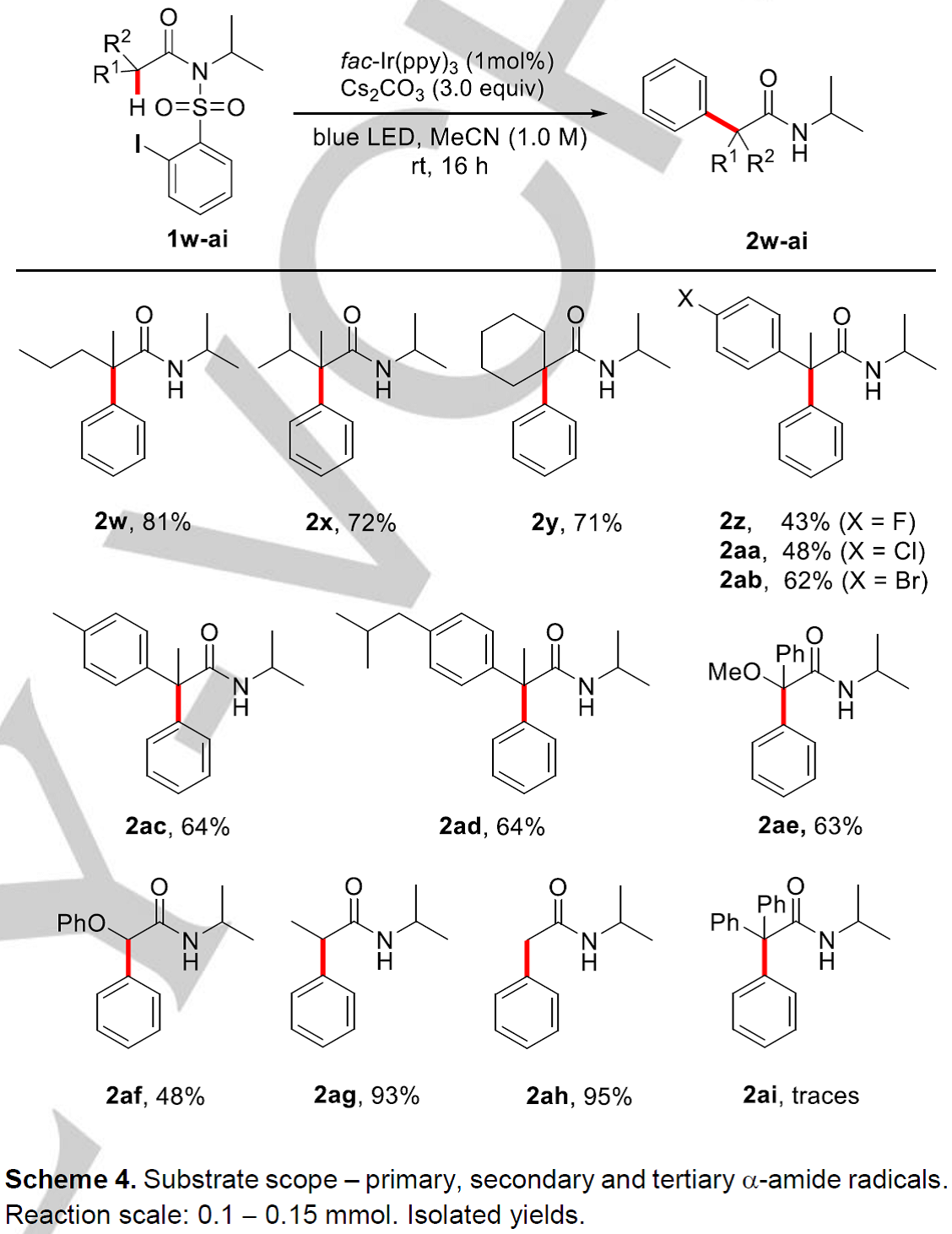
然后,作者在方法A的作用下对酰胺α-位不同取代基取代的范围也进行了探索(Scheme 4)。当把α-二甲基取代底物1a换成α-甲基正丙基(2w)、α-甲基异丙基(2x)、α-甲基芳基(2z-2ad)取代类底物后,反应均能顺利进行,以中等至良好的收率获得相应的α-C(sp3)-H键芳基化产物(43-81%);作者发现α-甲氧基取代类底物也能进行反应(2ae, 63%);除此之外,α-位为二级烷基和已经烷基时也能顺利进行反应(2af, 48%; 2ag, 93%; 2ah, 95%); 然而,α-二苯基取代类底物(2ai, traces)在该条件下不能适用。
机理研究
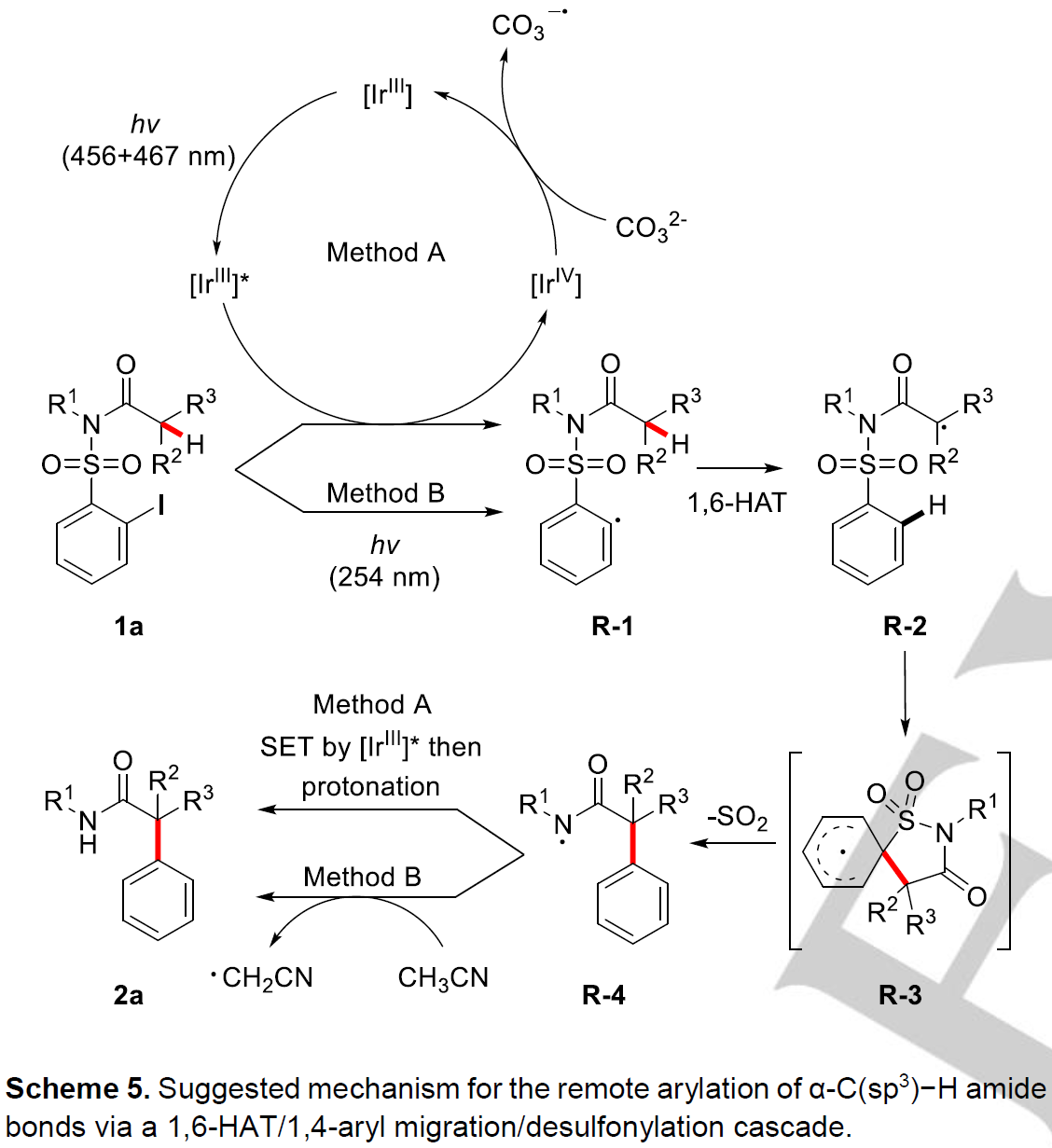
最后,作者对方法A和方法B可能的反应机理进行了探讨(Scheme 5)。对于Method A,首先,底物1a被激发态的[IrIII]*光催化剂还原生成芳基自由基R-1, 作者通过Stern-Volmer荧光猝灭实验证实了这一点;接着,R-1发生选择性的1,6-HAT生成自由基易位的α-酰胺自由基R-2; 然后,R-2发生本位进攻RTA-group的芳基部分生成环己二烯基自由基R-3; 接着发生芳构化,同时释放出SO2,生成酰胺自由基R-4; R-4被激发态的[IrIII]*催化剂还原,再被质子化生成目标产物2a。其中质子化过程中的质子来自于溶剂中存在的微量水,作者分别以氘代乙腈作为溶剂与加入氘代水进行氘代实验证实了该观点。[IrIII]*催化剂被氧化生成的[IrIV]物种与CO32-作用重新生成[IrIII]催化剂物种,这也解释了在该反应中需要当量的Cs2CO3的原因。此外,作者通过循环伏安法也证实了Cs2CO3与[IrIV]物种反应的的可能性。对于Method B,底物1a在紫外光辐射下C-I键直接均裂生成芳基自由基R-1,接着与Method A的过程相似,依次发生1,6-HAT、1,4-芳基迁移、释放出SO2生成自由基R-4; R-4再与反应溶剂乙腈发生氢原子转移生成目标产物2a, 同理,作者也通过氘代乙腈作为溶剂在Method B条件下进行反应证实了这一观点。
总结
德国明斯特大学Armido Studer教授课题组利用自由基易位芳基化(RTA)基团2-碘芳基砜基在可见光光催化条件下成功实现了N-烷基酰胺的α-C(sp3)-H键直接芳基化反应。利用该方法能够构筑全α-季碳中心酰胺化合物,同时避免了利用其他自由基芳基迁移策略构建α-季碳中心酰胺的底物局限性,底物无需进行预官能团化。
参考文献
[1] L. Desai, K. Stowers, M. S. Sanford, J. Am. Chem. Soc. 2008, 130, 13285. DOI: 10.1021/ja8045519 [2] J. He, M. Wasa, K.S.L. Chan, Q. Shao, J.-Q. Yu, Chem. Rev. 2017, 117, 8754 .DOI: 10.1021/acs.chemrev.6b00622 [3] S. M. Thullen, S. M. Treacy, T. Rovis, J. Am. Chem. Soc. 2019, 141, 14062. DOI:10.1021/jacs.9b07014 [4] D. P. Curran, D. Kim, H. T. Liu, W. Shen, J. Am. Chem. Soc. 1988, 110, 5900.DOI: 10.1021/ja00225a052 [5] X. Wu, C. Zhu, Acc. Chem. Res. 2020, 53, 1620.DOI: 10.1021/acs.accounts.0c00306 [6] F. Friese, C. Mück-Lichtenfeld, A. Studer, Nat. Commun. 2018, 9, 2808.DOI: 10.1038/s41467-018-05193-6 [7] Y. Li, B. Hu, W. Dong, X. Xie, J. Wan, Z. Zhang, J. Org. Chem. 2016, 81,7036.DOI: 10.1021/acs.joc.6b00735 [8] D. J. Leonard, J. W. Ward, J. Clayden, Nature 2018, 562, 105. DOI: 10.1038/s41586-018-0553-9













No comments yet.