

本文作者:杉杉
导读
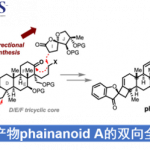
近日,康斯坦茨大学(Universität Konstanz)Tanja Gaich课题组在Angew. Chem. Int. Ed.上发表论文,报道了外消旋Waihoensene 以及对映体(+)-Waihoensene的全合成。(+)-Waihoensene作为二萜天然产物家族的成员之一,具有角三奎烷(angulartriquinane)的结构单元,同时四环[6.5.5.5]骨架均为顺式稠合,包含六个连续的立体中心,其中四个为季碳中心。此外,该全合成过程涉及非对映选择性自由基环化,分子内Pauson-Khand反应,非对映选择性α-烷基化和非对映选择性1,4-加成反应。
Total Synthesis of the Diterpene Waihoensene
Tanja Gaich, Lisa-Catherine Rosenbaum, and Maximilian Häfner
Angew. Chem. Int. Ed. ASAP DOI:10.1002/anie.202011298 https://doi.org/10.1002/ange.202011298

正文
含有聚奎烷(polyquinane)结构的萜烯天然产物构成了倍半萜,二萜和二倍半萜(sesterterpenes)中的多样化的结构,一直受到合成界的关注。与线性三奎烷3相比,角三奎烷(Angular triquinanes)2由于具有季碳中心导致合成上具有难度(Figure 1)。
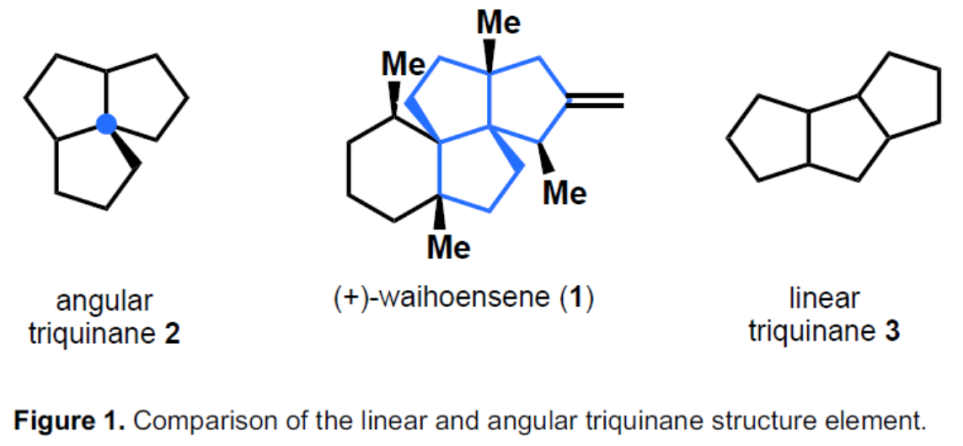
Waihoensene(1)具有角三奎烷的结构,且包含四个连续的季碳中心。1997年,Weavers等人[1]从新西兰罗汉松(Podocarpus totara var waihoensis)中分离出了(+)-Waihoensene。从结构上讲,它包含一个高度拥挤且顺式稠合的四环母核以及六个连续的立体中心,其中四个为连续的碳季中心。除了存在四个季碳立体中心以外,Waihoensene(1)合成还存在另一难题,由于大多数C-C键形成需要官能团,而其结构中除单个双键外不包含任何官能团,因此需对中间体过度功能化以及额外的去官能化过程,从而导致合成路线过长。
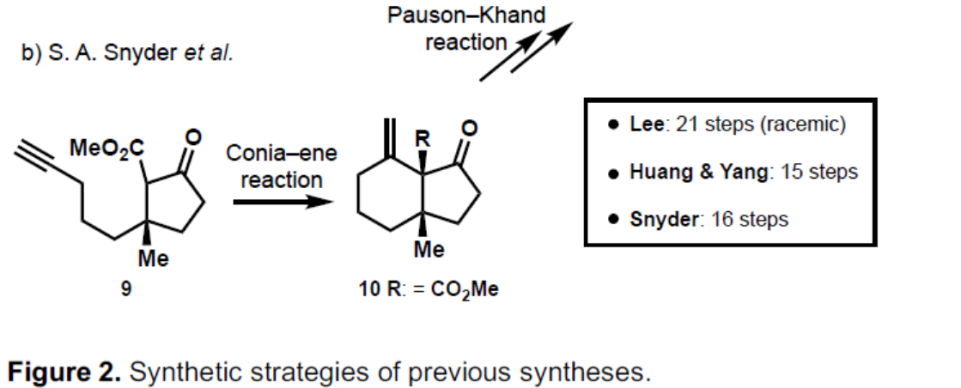
目前,已有三个课题组实现Waihoensene(1)的全合成(Figure 2)。2017年,Lee等人[2]首次实现了(±)-Waihoensene的全合成,其特征是串联的[2+3]环加成反应以构建具有两个连续季碳立体中心的三环。2020年,由Huang和Zhang[3]以及Snyder[4]课题组提出了涉及Conia-Ene/Pauson-Khand策略。后面两种合成方法都是对映选择性的,步数范围为15和16而,Lee的外消旋合成需要21个步骤。
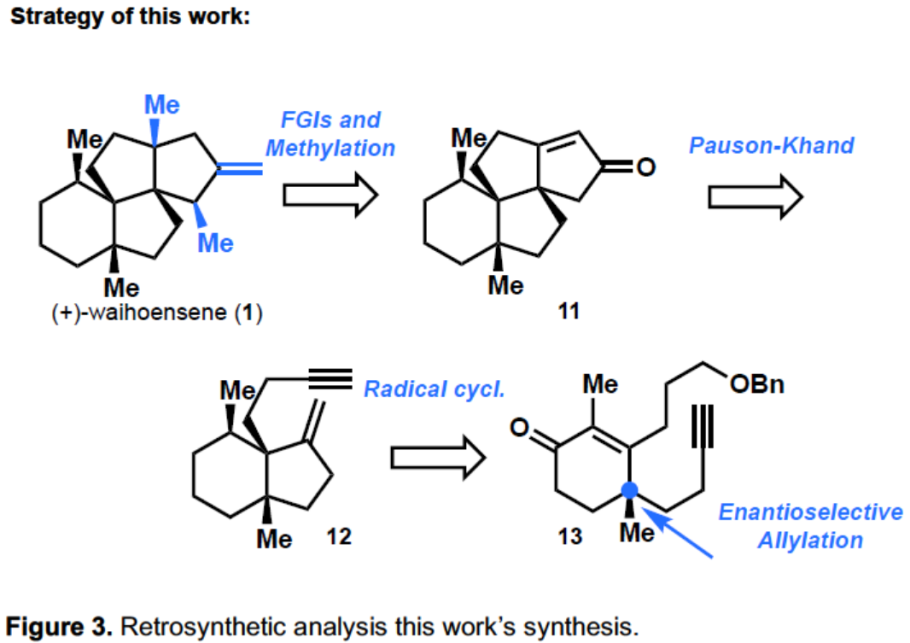
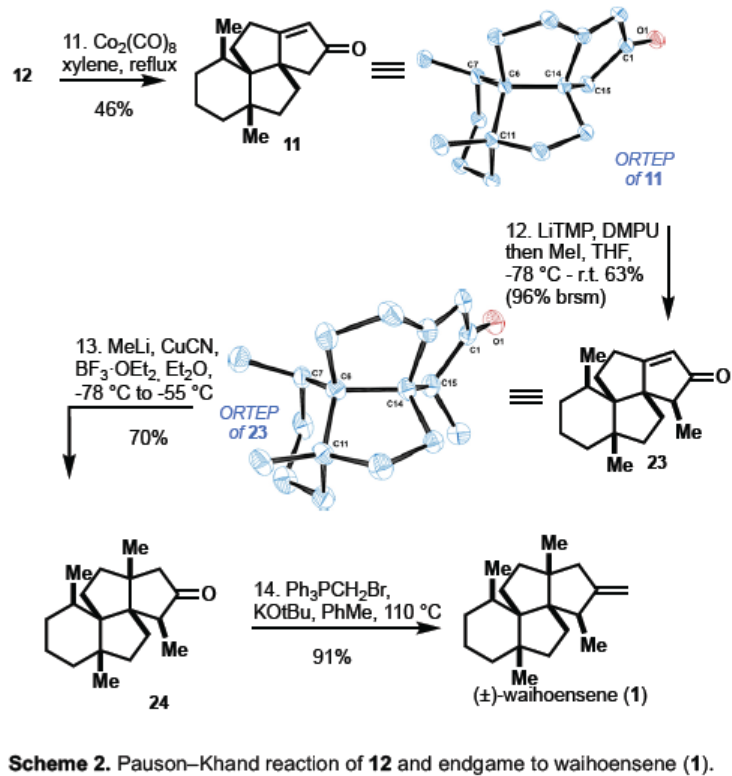
在此,康斯坦茨大学Tanja Gaich课题组报道了Waihoensene(1)的外消旋(14步)和对映选择性(19步)的合成。首先,通过不对称的烯丙基化反应构建具有四个季碳立体中心的化合物27(Scheme 3)。然后,通过自由基环化反应,来合成18中六氢茚满(hydrindane)而,所有其他合成方法都不会将这两个转化结合在一起。双环18作为Pauson-Khand反应的刚性模板,最终化合物11经三步转化从而合成Waihoensene(1)。
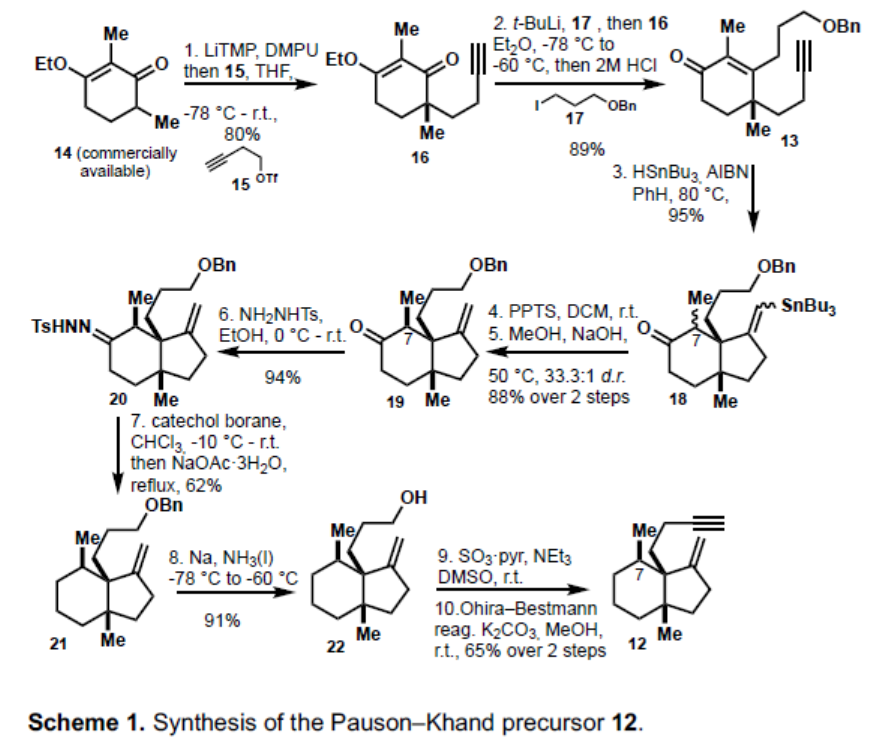
首先,以市售化合物14为初始底物(Scheme 1)。通过14与15的烷基化反应,可将季碳中心引入形成16。紧接着,将其加入到17中,原位消除反应得到烯酮13,经自由基环化反应,以95%的收率形成所需的茚满骨架18。18经脱甲锡烷基化反应(Destannylation)和在含有氢氧化钠的甲醇溶液中反应,得到所需构型的C-7甲基,以88%收率得到所需的全顺式19。随后,19中的酮与肼反应形成20,再将其还原得到化合物21,从而完成了19中酮的去官能化。通过在Birch条件下进行脱保护,将Parikh-Doering氧化为相应的醛以及Ohira-Bestmann烷基化反应,将其转化为炔12。
炔12作为Pauson-Khand反应的前体,可与Co2(CO)8反应形成相应的钴-炔配合物,经环化和共插入后,形成46%收率的四环11,其晶体结构进一步证明了结构的正确性(Scheme 2)。合成的最后阶段包括三步过程,从Pauson-Khand产物11的α-烷基化开始,得到单一非对映异构体23。与之前报道相反[2],即甲基与烯酮11的1,4-加成,随后的α-烷基化和最终的烯烃化,从而得到Waihoensene(1)。作者在进行11的α-烷基化反应时没有遇到任何困难,顺利获得产物23,其晶体结构进一步确定了产物的构型。值得注意的是,在α-烷基化反应中,碱和添加剂的选择对于动力学和热力学烯醇化物的竞争形成至关重要。使用强碱,即LDA,LiTMP和LiICA与DMPU结合,可得到所需的甲基化23作为唯一产物。使用较弱的碱(如LiHMDS),则形成热力学烯醇化物(烯酮部分的γ-位去质子化)。随后,将铜酸甲酯经1,4-加成到23中,以70%的收率得到24。最后,通过Wittig烯烃反应,以91%的收率获得天然产物Waihoensene(1)。
为了获得光学活性产物,作者决定通过对映选择性烯丙基化反应引入第一个季碳立体中心(Scheme 3)。以乙烯基类硫酯25为初始底物,用碘甲烷将25进行季铵化可得到26,钯催化并使用Trost’s配体,实现脱羧烯丙基化从而获得27,ee为96%,收率为87%。随后,将乙烯基硫代酸酯转化为相应的甲酯28,经氢硼化/氧化和Ohira-Bestman反应,形成30。最后,按照上述的即可获得(+)-Waihoensene(1)。值得注意的是,克级实验不会损失收率和光学活性。

总结
康斯坦茨大学Tanja Gaich课题组报道了(±)-Waihoensene(1)全合成最短的路线,仅需14步骤。而对于对映选择性路线,需要19步,并且获得30克的光学活性产物。该全合成过程涉及非对映选择性自由基环化,分子内Pauson-Khand反应,非对映选择性α-烷基化和非对映选择性1,4-加成反应。
参考文献
[1] D. B. Clarke, S. F. R. Hinkley, R. T. Weavers, Tetrahedron Lett. 1997, 38, 4297-4300.[2] H. Lee, T. Kang, H.-Y. Lee, Angew. Chem. Int. Ed. 2017, 56, 8254-8257; Angew. Chem. 2017, 129, 8366-8369.
[3] Y. Qu, Z. Wang, Z. Zhang, W. Zhang, J. Huang, and Z. Yang J. Am. Chem. Soc. 2020, 142, 6511-6515.
[4] C. Peng, P. Arya, Z. Zhou, and S. A. Snyder Angew. Chem. Int. Ed. 2020, 59, doi.org/10.1002/anie.202004177; Angew. Chem. 2020, 132, doi.org/10.1002/ange.202004177.
















No comments yet.