

投稿作者alberto-caeiro
在生物酶催化反应条件下,生物体内光学活性天然产物的存在并不特殊,但在一些手性天然产物的代谢产物中,经常会发现外消旋体的存在,这在当代谢途径是没有酶的存在条件下反应是可能的。这些外消旋体为合成化学家们提供了灵感,往往会为他们提供有效的合成路径。
1990年和1993年,Itokawa在中国和印度分别发现了一种药草钩毛茜草和茜草[1](图片来源见水印),并从其中分别分离出了天然产物Rubioncolin A, B和Rubicordifolin。这几个天然产物因其独特的二聚结构,引起了加州大学伯克利分校的D. Trauner教授的兴趣,并且从茜草中分离得到的都是外消旋体结构,这说明在其中存在无生物酶催化的反应。下图列出了其可能的生物合成机理。猜想的单体萘醌4经a途径瞬时的环化得到苯并呋喃5,随后立刻与4发生D-A反应,二聚得到rubioncolin A;单体萘醌4经过b途径形成环内酯7,另外的,苯并呋喃脱氢得到的二取代烯烃8,环内酯7与烯烃8经过hetero-D-A反应得到消旋体rubicordifolin;苯并呋喃5与6先酯化,在异构化形成α,β-不饱和酮,随后发生hetero-D-A反应得到rubioncolin B。
2005年和2008年,通过对仿生合成的研究,D. Trauner组完成了rubicoedifolin[2]和rubioncolin B[3]的全合成,并确认其结构。
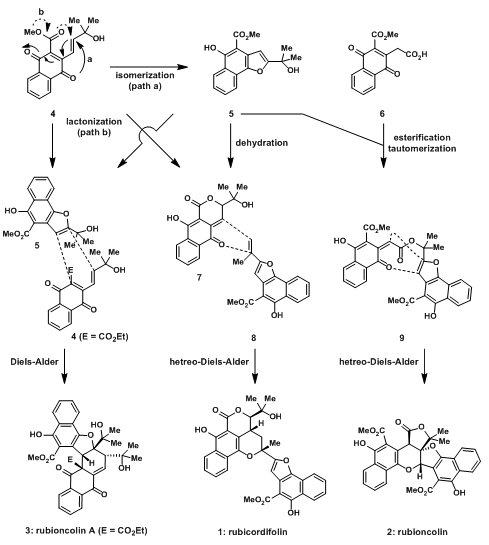
图1:Possible biosynthesis of rubioncolin A and B and rubicordifolin
Rubicordifolin全合成
- 逆合成分析
正如猜想的生物合成路线,rubicordifolin可以由7和8两个碎片经过D-A反应得到,两个碎片都可由设想的生物前体4得到。另外,在适宜的条件下,前体4可以在原位反应得到前体7和8,随后发生串联反应直接二聚得到产物;生物前体4可以由已经制备得到的10和11得到。
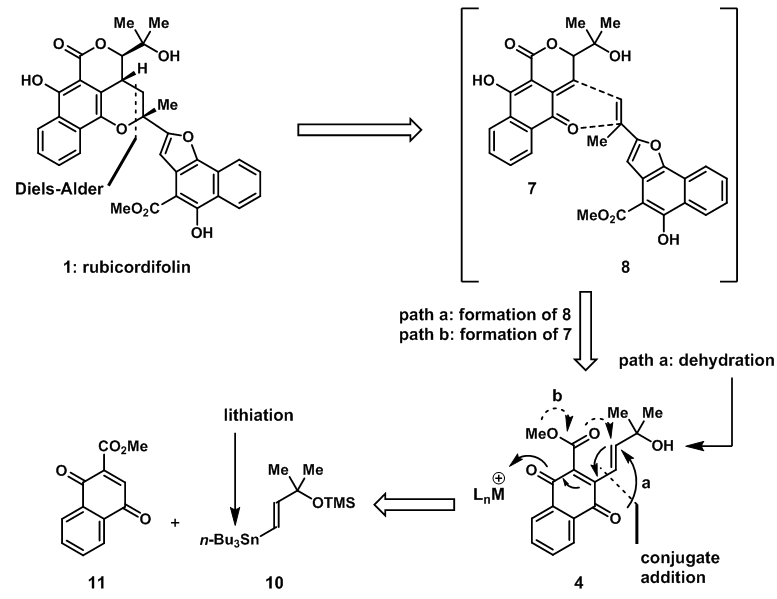
图2:Retrosynthetic analysis of rubicordifolin(1)
- rubicordifolin的全合成
D.Traunei教授的全合成工作从已经得到的底物11开始,11与烯基锡试剂10反应,再经过异构化得到12;用CAN氧化12得到生物前体4,生物前体4自发反应得到苯并呋喃衍生物5和14,羰基O作为亲核试剂发生Michael加成得到13,path a中,脱氢得到5,而在path b 中,消除丙酮得到furomollugin 14。
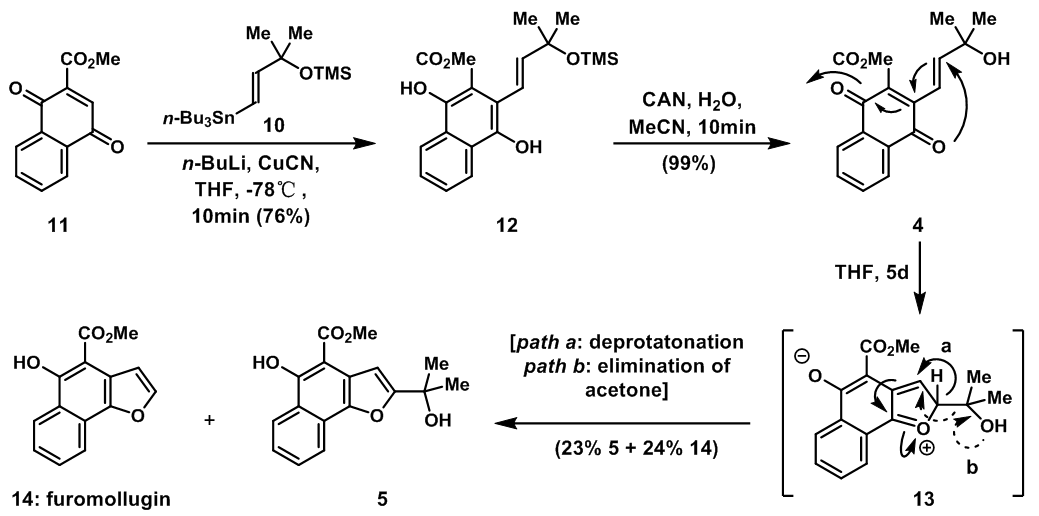
图3:Synthesis and spontaneous reaction of naphthoquinone 4
受到生物合成的启发,选择将生物前体4和苯并呋喃体系5在PbB(OH)2存在下加热发生分子间D-A反应得到rubioncolin A,但是并没有如愿观测到希望的产物。相反的,通过三级醇脱水得到的烯烃8为主要产物,此外还有少量的rubicordfolin。
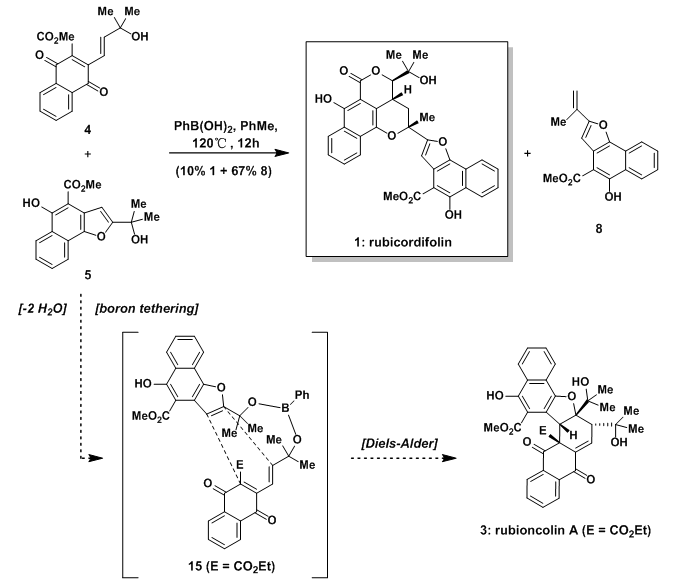
图4:Synthesis of rubicordifolin during an attempted preparation of rubioncolin A
随后的机理实验研究为rubicordfolin的形成和合成提供了合理的解释并提升了其产率。
因为4生成苯并呋喃体系5的反应是自发的,这可以作为串联反应形成rubicordfolin。将前体4置于PbB(OH)2中,在120℃条件下反应,以45%收率得到rubicordfolin。在Lewis酸催化下,4经path a形成16,16经path a脱水得到17,经path b的带furomollugin 14;另一方面,在Lewis酸催化下,4发生环内酯化得到18;17脱水,18脱除甲醇分别得到20和19,两者发生仿生的杂D-A反应,得到了天然产物rubicordfolin。

图5:Optimized cascade synthesis of rubicordifolin
Rubioncolin B 全合成
- 逆合成分析
D.Trauner教授对于中间的二氢吡喃环准备采用的是分子内的D-A反应,于是前体21经D-A反应后,脱除甲基即可得到天然产物;21是22的异构体,而22可以通过23氧化得到;酯化反应可以将碎片24和25合成得23;14可以通过金属锂试剂与丙酮发生亲和加成反应得到;而14与23都可由原料11制得。
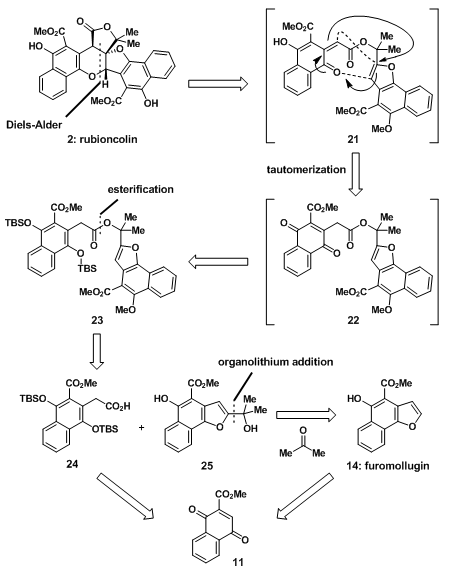
图6:Retrosynthetic analysis of rubioncolin B
2.rubioncolin B的全合成
虽然在之前的合成中已有方法由11得到14,但在mollugin的全合成中,有一种新的合成14的方法得到应用。原料11首先与烷基锡试剂发生Sakurai-type 烷基化反应,然后将形成的双羟基与碘化钾反应得26;随后在Grubbs Ⅱ催化剂催化下发生烯烃复分解反应得到27;27被CAN氧化得到28后,将其与三乙胺混合,碱性条件下脱氢得到中间体29,发生6Π电环化对旋得到mollugin30;30再被CAN氧化得到半缩酮31,同样将31与碱碳酸钾混合,发生串联反应得到14,然后用甲基保护羟基得到甲氧基保护产物33;随后有机锂试剂与丙酮反应得到25。
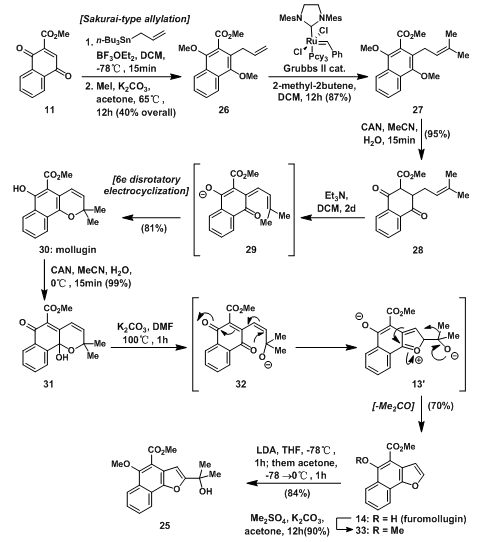
图7:Synthesis of tertiary hydroxyl fragment 25
羧酸碎片24的合成中,也使将原料11与烷基锡试剂经Sakarai-type烷基化反应后,用TBSCl与羟基反应得到34;随后的两步氧化反应将双键氧化成羧基得到碎片24,24接着与草酰氯反应得到相应的酰氯35;然后35与25在三乙胺催化下发生反应得到D-A反应前体23。
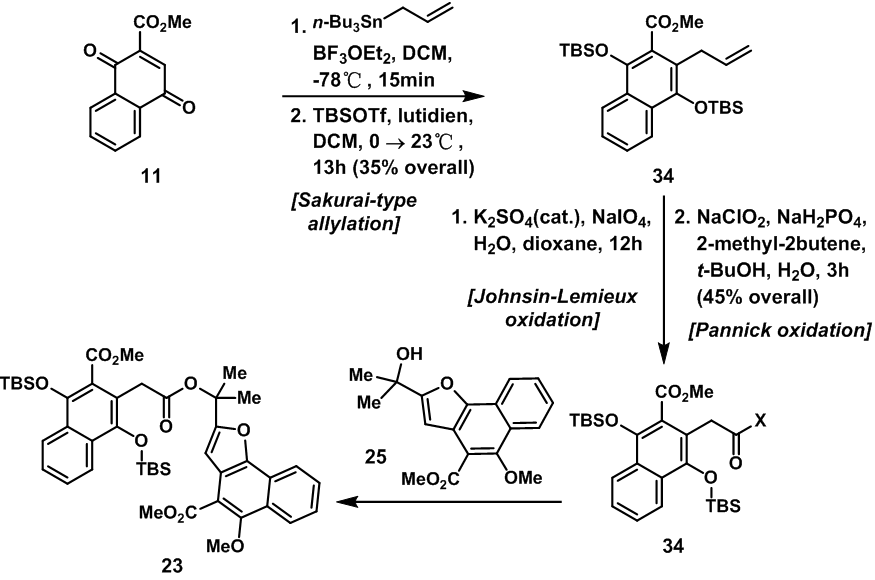
图8:Synthesis and coupling of carboxylic acid 24
D-A反应前体酯23在PhI(OAc)2和TAS-F的存在下发生反应,以60%的收率得到38;23先脱除TBS保护得到的36,在PhI(OAc)2和F–作用下脱除另一TBS保护基得到萘醌22,22异构化得到21后,发生endo-D-A反应,最终得到38;在三溴化硼作用下,38脱除甲基得到天然产物rubioncolin B。

图9:Final stage and completion of the total synthesis of rubioncolin B
总结
通过对天然产物生物起源的分析,科研人员可从中收到启发,完成精美而优雅的全合成工作,D. Trauner教授的全合成工作和其他许多经典的全合成工作一道,再次向世人展现了自然界的神秘和精美!
在本次的全合成工作中,D-A反应也得到了关键的应用,这也说明了D-A反应在化学反应中的重要作用!
参考文献
- a) Y.-F. Qiao, K. Takeya, H. Itokawa, Y. Iitaka, Pharm. Bull. 1990, 38, 2896; b) H. Itokawa, Z. Z. Ibrahemi, Y.-F. Qiao, K. Takeya, Chem. Pharm. Bull. 1993, 41, 1869;
- -P. Lumb, D. Trauner, J. Am. Chem. Soc., 2005, 127, 2870. DOI: 10.1021/ja042375g;
- -P. Lumb, K. C. Choong, D. Trauner, J. Am. Chem. Soc., 2008, 130, 9230. DOI: 10.1021/ja803498r;
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!






















No comments yet.