
作者:杉杉
导读:
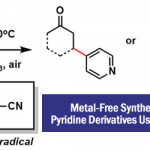
近日,中国科学技术大学与淮北师范大学的黄汉民课题组在Angew. Chem. Int. Ed.中发表论文,报道一种全新的I2/Ni-或BnX/Ni-催化aminodienes的分子内环化carboamination反应方法学,进而成功完成N-杂环分子的构建,涉及C-N键活化的过程。
Intramolecular Carboamination of Aminodienes to N-Heterocycles via C−N Bond Activation
X. Yan, B. Yu, H. Liu, H. Huang, Angew. Chem. Int. Ed. 2024, ASAP. doi: 10.1002/anie.202316563.
正文:
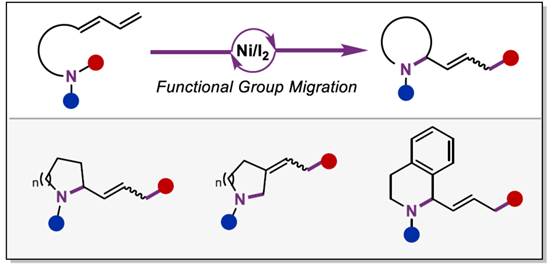
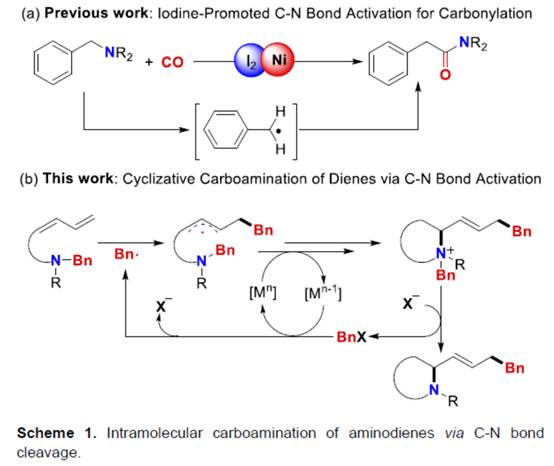
近年来,诸多研究团队已经成功设计出多种aminodienes参与的分子内氢胺化反应方法学[1],涉及在C=C键上进行N-H键加成的过程,是构建N-杂环分子的有效策略。然而,对于aminodienes参与分子内的carboamination反应方法学,目前仍有待进一步的研究报道。受到近年来通过碘促进C-N键活化参与羰基化反应方法学[2] (Scheme 1a)以及季铵盐的C-N键易被卤素阴离子进行断裂[2]的相关研究报道的启发,这里,中国科学技术大学与淮北师范大学的黄汉民课题组报道一种全新的I2/Ni-或BnX/Ni-催化aminodienes的分子内环化carboamination反应方法学,进而成功完成N-杂环分子的构建 (Scheme 1b)。
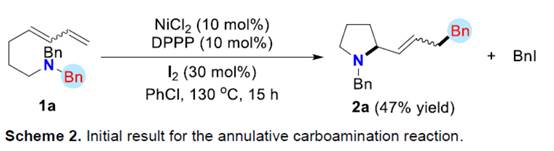
首先,作者采用aminodienes 1a与BnCl作为模型底物,进行相关反应条件的尝试(Scheme 2)以及优化筛选 (Table 1)。进而确定最佳的反应条件为:采用NiCl2作为催化剂,DPPP作为配体,在PhOMe反应溶剂中,反应温度为130oC,最终获得79%收率的产物2a (Z/E为8:1)。
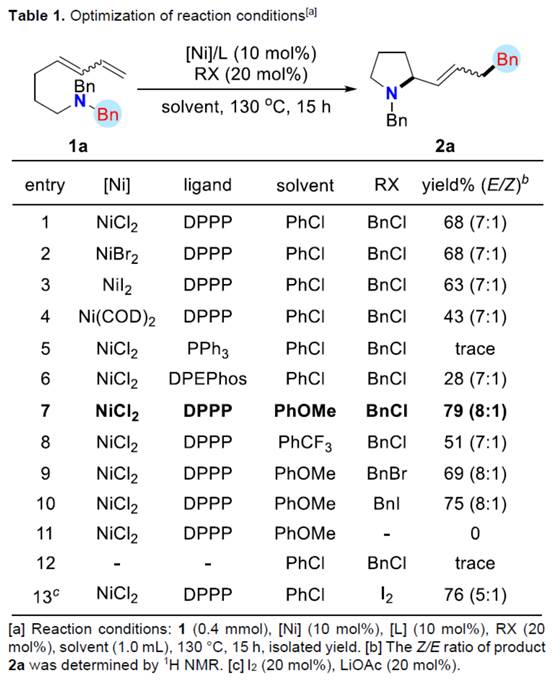
在上述的最佳反应条件下,作者对合成吡咯烷衍生物的底物 (Table 2)的应用适用范围进行深入研究。
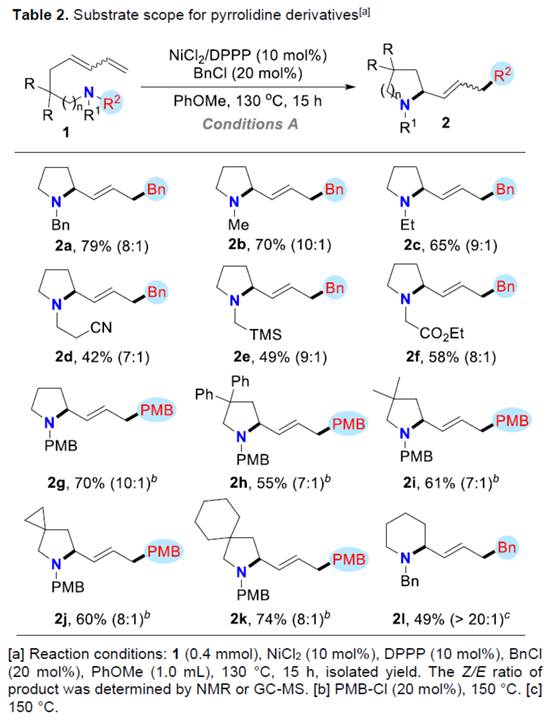

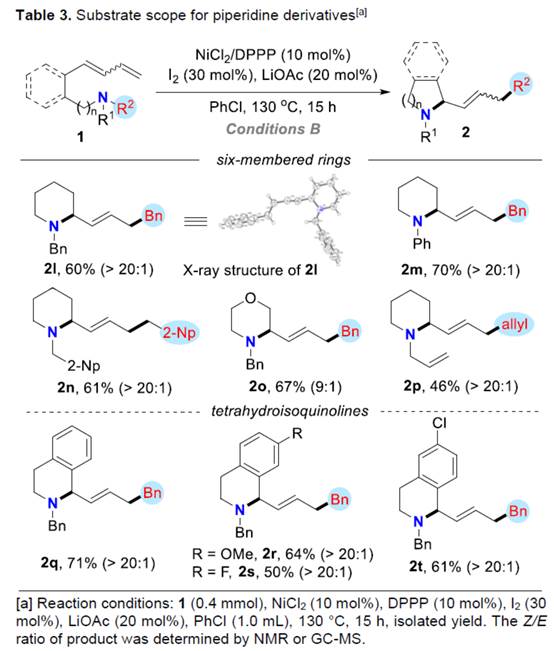
同时,通过对反应条件的优化,作者还对合成哌啶衍生物的底物 (Table 3)的应用适用范围进行深入研究。

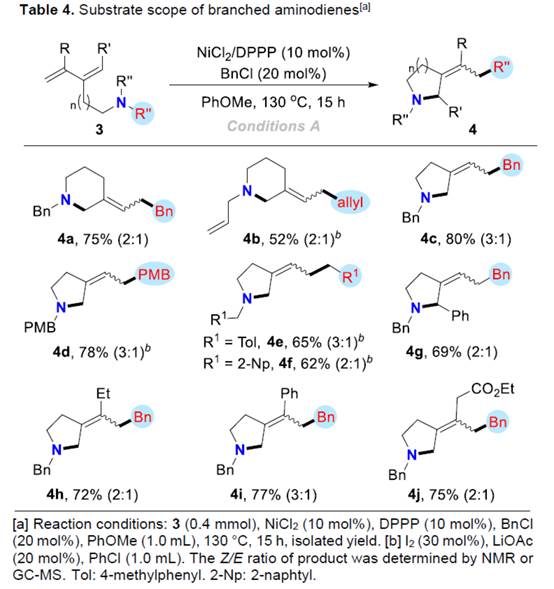
之后,该小组还对支链类型的aminodienes底物 (Table 4)的应用范围进行深入研究。

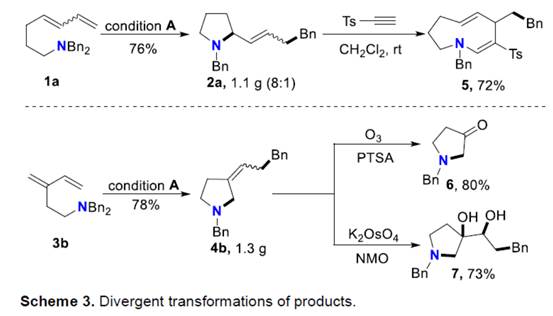
之后,该小组通过如下的一系列研究进一步表明,这一全新的分子内carboamination策略具有潜在的合成应用价值 (Scheme 3)。
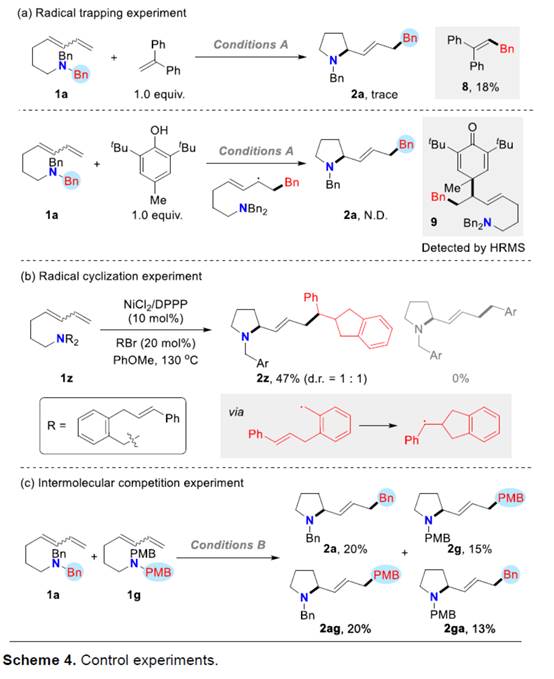
接下来,作者对上述分子内carboamination过程的反应机理进行进一步研究 (Scheme 4)。

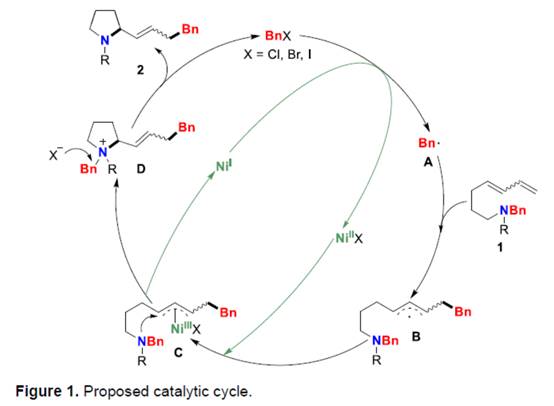
基于上述的实验研究以及前期相关的文献报道[1],作者提出如下合理的反应机理 (Figure 1)。
总结:中国科学技术大学与淮北师范大学的黄汉民课题组报道一种全新的I2/Ni-或BnX/Ni-催化aminodienes的分子内环化carboamination反应方法学,进而成功完成N-杂环分子的构建,涉及C-N键的活化。这一全新的carboamination合成转化策略具有底物范围广泛、优良的官能团兼容性以及优良的原子经济性等优势。
参考文献:
[1] H. Yu, B. Gao, B. Hu, H. Huang, Org. Lett. 2017, 19, 3520. doi: 10.1021/acs.orglett.7b01488.
[2] H. C. Brown, K. L. Nelson, J. Am. Chem. Soc. 1953, 75, 24. doi: 10.1021/ja01097a007.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.














No comments yet.