
本文作者:杉杉
导读
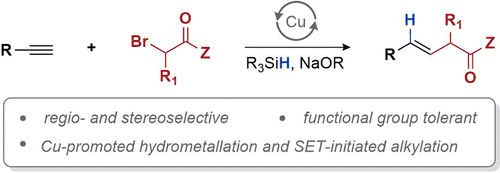
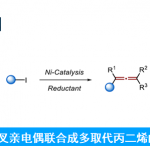
近日,美国Washington大学 (University of Washington)的G. Lalic教授课题组报道了端炔与α-溴代羰基化合物之间的立体选择性偶联反应方法学,从而获得具有高度E-选择性的烯基化合物。该方法学具有良好的官能团兼容性与广泛的底物适用范围。同时,反应机理研究表明,α-溴酸酯对烯基铜中间体的SET氧化过程为偶联过程的关键步骤。
Hydroalkylation of Alkynes: Functionalization of the Alkenyl Copper Intermediate through Single Electron Transfer Chemistry
A. Hazra, J. Kephart, A.Velian, G. Lalic, J. Am. Chem. Soc. ASAP. doi: 10.1021/jacs.1c03396.
正文
炔烃的氢烷基化反应 (hydroalkylation)作为构建烯基化合物的有效策略之一,能够更好地实现未活化的烷基亲电试剂与炔基化合物之间的直接偶联过程[1]-[8]。在过去几年中,已经开发出多种具有高度区域与立体选择性氢烷基化反应方法学,从而能够获得三种不同类型的双取代烯基化合物,即1,1-二取代烯基化合物、Z-烯基化合物或E-烯基化合物 (Scheme 1a)。尽管氢烷基化方法学的研究已经取得较大进展,然而,这一方法学对于部分官能团化的偶联参与物 (coupling partner)而言,实现其相应的选择性氢烷基化过程目前仍面临较大的挑战,尤其对于α-卤代羰基化合物。这一类型的亲电试剂具有较高的氧化还原电势,并且,在各类过渡金属试剂存在下,能够较为容易地通过SET过程,产生相应的烷基自由基[9]-[10]。然而,通过SET过程实现选择性的加氢烷基化,则面临较多的挑战。目前为止,对于氢烷基化策略的研究主要涉及通过SET过程产生的烷基自由基与炔基化合物之间的直接加成过程 (Scheme 1b)。然而,上述的直接加成过程,反应速率较为缓慢[11]-[12],并且,通常形成E/Z两种异构体的混合物[11], [13]-[15]。迄今为止,直接的自由基加成方法学通常仅能够成功应用于活化的芳基取代型炔基化合物[16],并能够获得具有良好Z-立体选择性的烯基化合物。
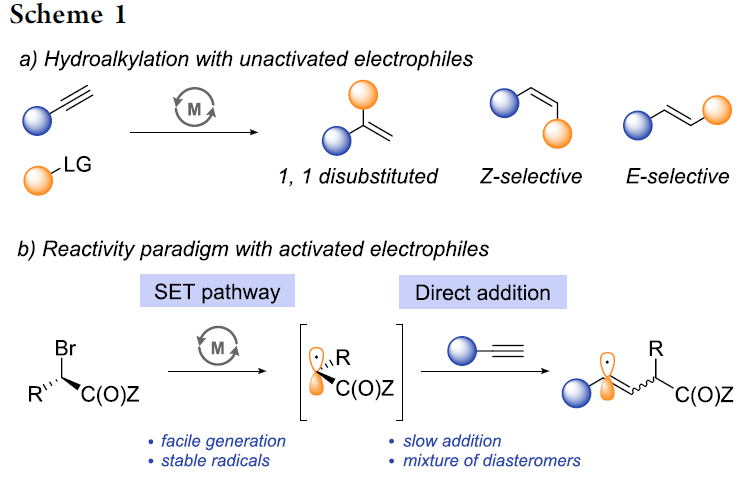
同时,近期在铜氢化物化学 (copper hydride chemistry) 的研究表明,通过炔基化合物铜氢化 (hydrocupration)过程产生的E-烯基铜 (E-alkenyl copper)中间体进行的后续官能团化过程,能够成功完成一系列炔基化合物氢官能团化反应 (hydrofunctionalization) [17]-[25]。基于上述文献报道,作者设想,能否将炔基化合物的铜氢化过程与α-卤代羰基化合物的SET化学进行结合,从而能够有效地完成具有良好区域与立体选择性的氢烷基化反应 (Scheme 2)。首先,通过高度区域选择性与立体选择性的铜氢化步骤,能够获得具有优良E-选择性的anti-Markovnikov加成产物,即E-烯基铜中间体。随后,通过ISET (inner-sphere SET)途径,进行卤原子转移 (halogen atom transfer)步骤,使烯基铜中间体进一步进行后续的烷基化过程。通过将上述两步过程的巧妙结合,作者最终成功开发出一种全新的炔基化合物氢烷基化策略。
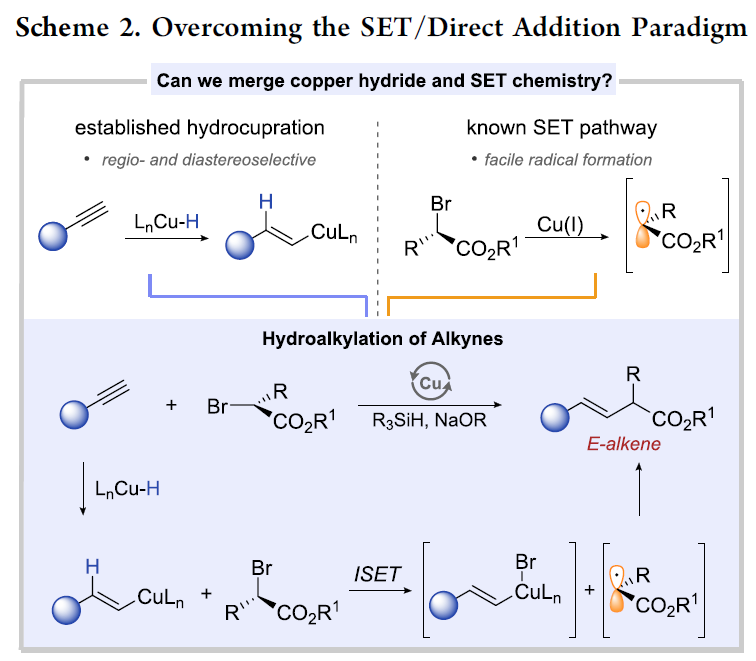
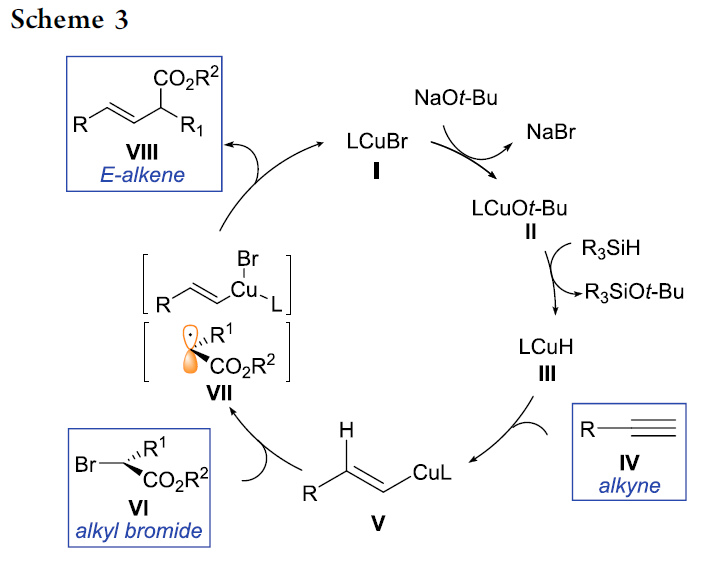
接下来,作者对于上述设想的氢烷基化过程提出一种可能的反应机理 (Scheme 3)。首先,LCuBr与t-BuONa以及R3SiH作用,形成LCuH配合物 III。随后,III与炔基化合物 IV反应,产生关键的烯基铜中间体 V,接下来,V与烷基溴底物 VI经历ISET过程,形成烯基铜(II)配合物以及碳中心自由基 VII。最终,烯基铜(II)配合物与碳中心自由基 VII通过自由基捕获 (radical capture)以及后续的还原消除过程,产生目标产物 VIII。
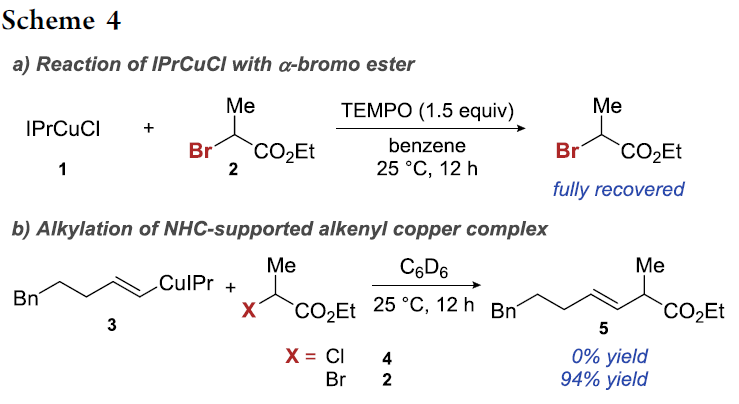
与此同时,作者进一步设想,通过NHC配体的配位,能够有效地调节上述烯基铜中间体的氧化电势,进而顺利实现上述设想的加氢烷基化过程。并且,已有文献报道,采用NHC配体,例如IPr,通常有利于形成线性的二配位Cu(I)配合物[26]-[29]。同时,研究发现这种线性的二配位Cu(I)配合物具有优良的氧化抑制性能,即在空气中能够表现出优良的稳定性。由此作者推测,通过NHC配体的σ-供体性能,能够产生具有较高还原电势的Cu(I)中间体,这类NHC配位的Cu(I)中间体在与α-卤代羰基化合物进行反应时,表现出较低的反应速率[30]-[31]。此外,作者同样观察到通过IPr配体的配位,能够加速铜氢化过程的进行,进而促进烯基铜中间体的产生。因此,上述将炔基化合物的铜氢化过程与α-卤代羰基化合物的SET化学进行结合的设想具有可行性。同时,作者对于加氢官能团化反应中常用催化剂IPrCuCl进行相应氧化还原电势的测定 (详见Supporting Information)。结果表明,其阳极峰电势 (anodic peak potential)高于其它各种类型的Cu(I)催化剂[32]。因此,IPrCuCl与α-卤代羰基化合物之间的反应过程较难进行。接下来,作者在室温条件下,将二级α-溴代酯 2加入至IPrCuCl (1)与TEMPO体系中时,发现并未观察到α-溴代酯 2参与相关的反应过程。这一结果表明反应步骤中无SET过程的发生 (Scheme 4a)。同时,作者进一步观察到,烯基铜配合物 3在类似的反应条件下,明显更易发生氧化,并能够获得交叉偶联产物 5 (Scheme 4b)。而对于反应性较低的亲电底物,例如,α-氯代酯4,则无法获得预期的偶联产物。
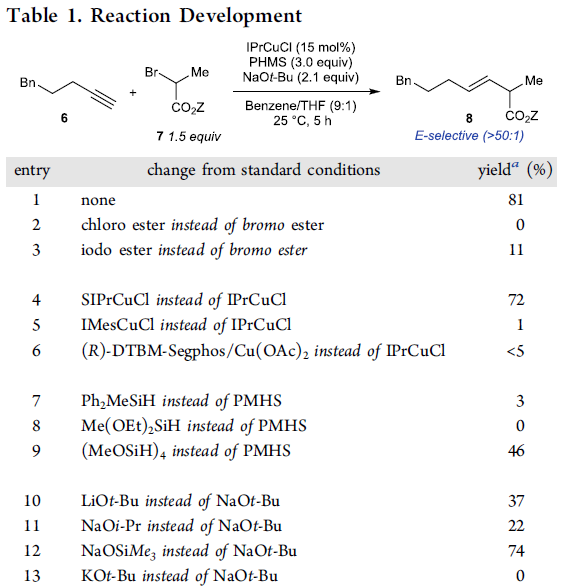
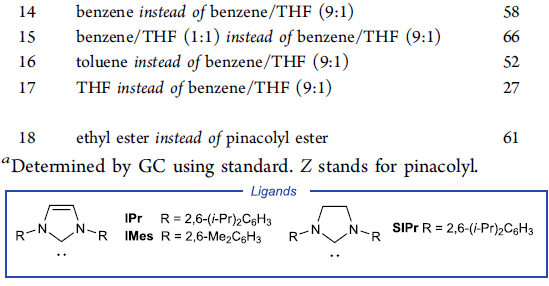
受到上述实验结果的鼓舞,作者采用端炔6与α-溴代酯7作为模型底物,进行相关偶联反应条件的优化筛选 (Table 1)。研究表明,选择IPrCuCl作为催化剂,聚甲基氢硅氧烷 (polymethylhydrosiloxane, PMHS)作为氢负离子源 (hydride source),t-BuONa作为转化试剂 (turnover reagent),苯/THF作为反应溶剂,反应温度为25 oC,最终能够获得81%收率的偶联产物8。
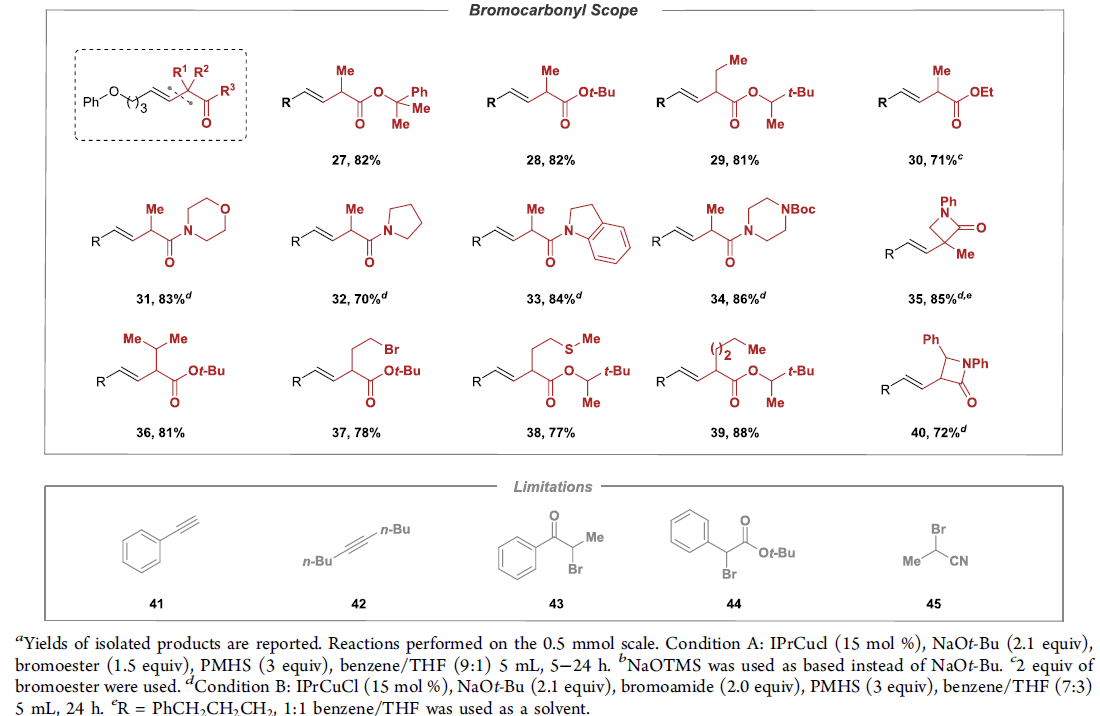
在获得相应的最佳反应条件之后,作者开始对上述偶联反应的底物应用范围进行考察 (Table 2)。研究表明,一系列带有不同取代基,例如烷基、环已基、杂芳基等的端炔底物,均能够顺利参与上述的偶联过程,并获得相应的E-烯基化合物8–26,其中,反应过程的立体选择性均大于50:1。接下来,作者发现,一系列二级α-溴代酯底物均能够与上述最佳反应条件良好地兼容,并获得相应的偶联产物27–30以及36–39。此外,作者进一步研究发现,通过调节相关反应条件中的溶剂配比与试剂加入计量,并延长反应时间,同样能够将二级α-溴代酰胺底物应用于上述偶联过程,进而获得相应偶联产物31–35以及40。同时,该小组观察到,上述偶联反应方法学同样存在一定局限性,即上述反应条件对于芳乙炔 (41)、二取代内炔 (42)、α-溴代酮 (43)、α-溴代腈 (45)、芳基取代的α-溴代酯 (44)、一级α-卤代羰基化合物、带有供质子性官能团 (protic functional groups,例如羟基,氨基)以及带有可还原性官能团 (reducible functional group,例如醛基与酮羰基)的相关底物,均无法有效地参与上述偶联转化过程。

接下来,作者通过自由基捕获实验 (radical trap experiment),对反应过程中存在的SET步骤进行进一步验证 (Scheme 5)。在前期的研究中,已经表明,TEMPO的加入对于炔基铜中间体的亲电官能团化过程无显著影响[18]。然而,SET过程对于TEMPO的加入,却能够表现出一定程度的敏感性。作者在自由基捕获实验过程中观察到,向端炔 6与α-溴代酯 7的反应体系中加入20% mol的TEMPO,反应受到一定程度的影响,偶联产物的收率显著降低。而在加入150% mol的TEMPO时,则能够观察到上述的偶联过程完全受到抑制,无烯基化合物产生 (Scheme 5)。同时,作者通过烯基铜IPr配合物 3的计量学实验 (stochiometric experiment)研究表明,在3与α-溴代酯 2的反应中,加入2eq. 的TEMPO时,能够获得63%收率的TEMPO加成产物48,进而证实TEMPO的加入,能够有效地抑制反应过程中的交叉偶联步骤。

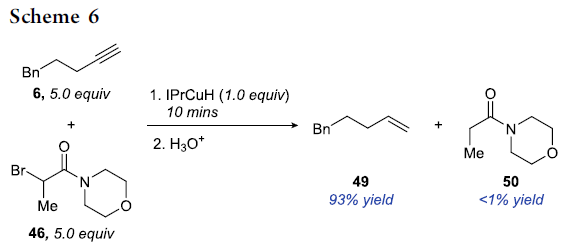
此外,由于IPrCuH与烯基铜中间体相比,具有更为显著的还原性,即IPrCuH与α-溴代羰基化合物能够更为迅速地进行相应的SET过程。然而,在竞争实验中,作者观察到IPrCuH与端炔反应的速率却显著快于IPrCuH与α-溴代酰胺46反应的速率。而且,在竞争实验中,作者发现,能够趋近专一性地获得烯基化合物49。这可能源自于后处理过程中水的加入对烯基铜中间体 (3)的质子化 (Scheme 6)。同时,作者进一步发现,IPrCuH与α-溴代酰胺46的化学计量反应速率较慢,同时,TEMPO的加入无法抑制这一过程的进行,这一事实表明IPrCuH与α-溴代酰胺之间的反应过程,并未涉及SET步骤或经历自由基中间体 (详见Supporting Information)。综上结果表明,尽管IPrCuH具有极高的还原性,然而却能够显著地阻碍SET过程的进行。
总结
美国Washington大学的G. Lalic教授课题组报道了通过α-溴代羰基化合物作为烷基化试剂参与的端炔底物的氢烷基化反应方法学,从而获得具有高度立体选择性的E-烯基化合物。该方法学具有良好的官能团兼容性以及广泛的底物适用范围。反应机理研究表明,通过烯基铜中间体参与的直接烷基化偶联过程通过SET途径进行。



















No comments yet.