
本文作者:杉杉
导读
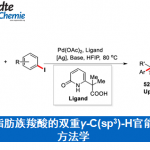
酮类官能团在有机化学中具有独特的反应性,可参与多种反应。同时,对于酮的催化合成方法也在不断地发展。近日,南京大学谢劲课题组在自然通讯(Nature Communications)杂志上发表论文,报道了通过光氧化还原、镍催化和正膦基自由基的协同作用实现芳基酸和芳基/烷基溴化物的亲电偶联反应。该方法可从廉价易得的底物开始,可直接合成具有高度官能化的酮衍生物,无需制备活化的羰基中间体或有机金属化合物,同时也作为常规Weinreb酮合成的补充。此外,使用合适的光催化剂、配体量和溶剂可以匹配上任何简单催化循环所需的反应速率。
Upgrading Ketone Synthesis Direct from Carboxylic Acids and Organohalides
Rehanguli Ruzi, Kai Liu, Chengjian Zhu,& JinXie
Nat.Commun. ASAP DOI: 10.1038/s41467-020-17224-2 https://www.nature.com/articles/s41467-020-17224-2
正文
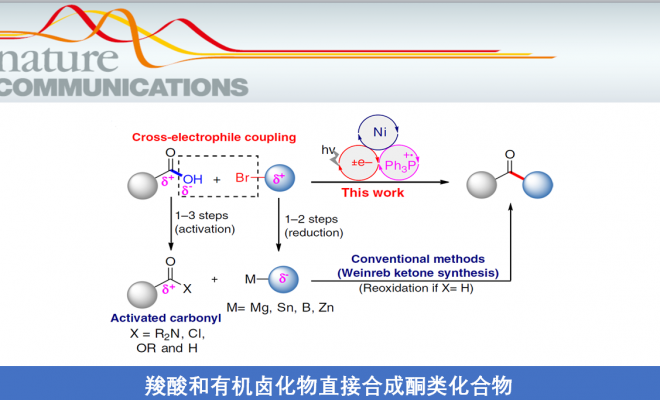
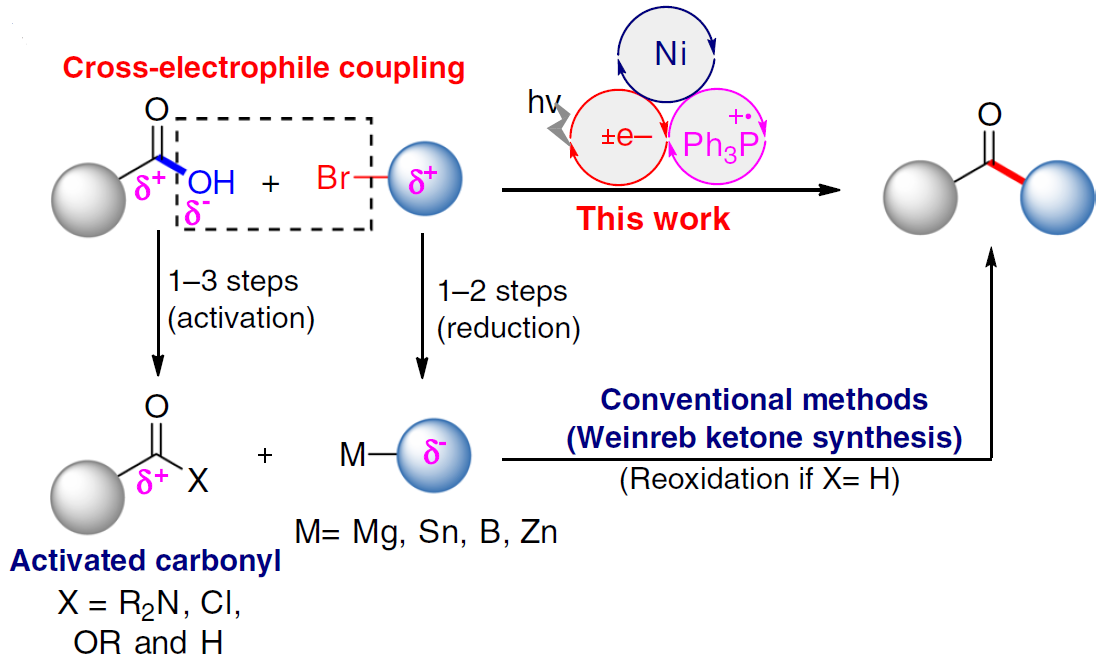
酮类化合物在有机化学中具有重要的作用,广泛存在于天然产物、药物、染料、香料等中。同时,也参与各类有机反应,如Mannich反应、Wittig 反应、Grignard反应、Passerini反应、Baeyer–Villiger氧化反应等。长期以来,开发一种从可从简单易得的底物直接合成酮类化合物的实用途径一直备受关注。羧酸和有机卤化物商业来源很丰富,并且结构多样且稳定性,常用于各类有机反应中(Fig. 1a)。由羧酸和有机卤化物合成酮时,常需要制备必要的中间体,如酰胺、醛或格氏试剂等(若使用醛,则必须进行再氧化)。此外,以催化的方法来生产酮类化合物时,往往通过过渡金属催化,实现活化的羰基(如酰基氯、酸酐)与有机金属试剂之间的碳-碳偶联(Fig. 1b)。然而,由羧酸活化的羰基通常多达三步制备,再与有机金属化合物(有机卤化物的金属化)反应,从而导致较差的官能团相容性以及存在冗长的保护/脱保护过程。
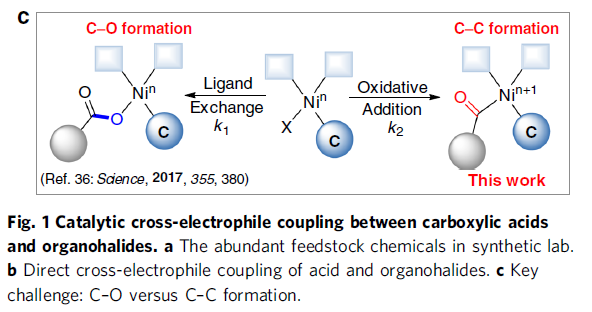
近年来,镍催化亲电偶联反应已引起了关注。在化学计量还原剂存在的条件下,可实现两种不同的亲电试剂的碳-碳偶联。作者假设,在交叉偶联反应中,羧酸可直接作为潜在的亲电试剂(C-末端),而不是亲核试剂(O-末端)。若实现此类偶联,则可直接简化羧酸和有机卤化物合成酮的过程。最近,谢劲课题组还报道了一种优雅的光氧化还原促进了羧酸脱氧的过程,从而产生了酰基自由基。据文献报道,通过光氧化还原和镍催化的组合,可实现羧酸与芳基溴化物的亲电偶联反应,而光氧化还原产生的酰基自由基的氧化加成过程,对抑制C-O键形成至关重要(Fig. 1c)。此外,Gong课题组也报道了通过将羧酸原位转化为酸酐以抑制C-O偶联。然而,由于C-O键的强解离能(106 kcal mol-1),在氧化加成步骤中使用游离羧酸作为酰基自由基前体仍是一个挑战。
由此,作者提出了一种可能的反应机理(Fig. 2)。首先,光氧化还原产生的三苯基膦自由基阳离子(I)与羧酸根阴离子重组形成正膦基自由基中间体(II)。由于正膦基自由基与氧之间的强亲和力,导致中间体II易发生β-断裂,形成关键的亲核酰基自由基。随后,酰基自由基与芳基-NiII中间体(III)进行氧化加成,形成NiIII配合物(IV)。最后,NiIII中间体(IV)经还原消除,即可生成所需的亲电偶联产物R-CO-Ar。同时,形成的NiI经SET从而获得IrII,并完成催化循环。此外,该亲电偶联反应中仍存在一些挑战:(1)有效控制C-O键断裂和镍中心自由基的加成;(2)减弱化学计量的三苯基膦对镍催化单元的干扰;(3)使用适当的配体和溶剂来显着抑制C-O键的形成。
随后,作者以4-甲基苯甲酸(1)与5-溴-2-(三氟甲基)吡啶(2)作为模型底物进行了反应条件的筛选(Table 1)。筛选结果表明,当使用2 mol%的[Ir{dF(CF3)ppy}2{dtbbpy}]PF6, 3mol%的NiBr2•dme,5 mol%的4,4′-二叔丁基-2,2′-联吡啶配体 (L1),1.5当量的Ph3P,可在DMF-CH3CN的混合溶剂中室温反应,获得82%收率的偶联产物(3),而C-O偶联产物3a’的收率为16%。再对其它条件的筛选时,如配体、溶剂等,均未提高收率。同时,作者发现配体使用量对于3与3a’的比例影响很大,而使用混合的碱和混合溶剂可改善羧酸的去质子化过程以加速酰基自由基的生成,并且使用一定量的配体将促进酰基自由基氧化加成到芳基镍(II)物质中,而增加或减少用量均不利于反应进行。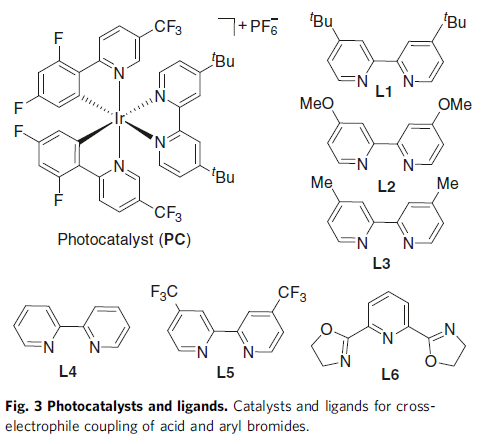
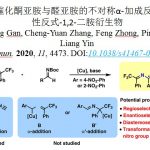
在获得上述最佳反应条件之后,作者开始对羧酸底物范围进行了扩展(Fig. 4)。反应结果表明,苯甲酸中的苯基取代不受电子效应和定位效应的影响,均可获得62-83%收率的产物3-21。同时,一些具有活性的官能团也能够很好的耐受,如溴(6),羰基(13-16),烯烃(18),炔烃(19)和乙缩醛(21)等,这些官能团中有一些很难通过常规的Weinreb酮合成方法获得。此外,杂芳基羧酸也取得令人满意结果,获得具有价值的二杂芳基酮衍生物22-26。然而,使用脂肪族羧酸时,只获得少量的所需产物,同时也发生脱羧性C-C偶联和C-O偶联反应。
![]()
随后,作者对芳基/烷基溴化物的底物范围进行了扩展(Fig. 5)。反应结果表明,一些市售的芳基溴化物均可以良好的收率获得所需产物27-46。同时,该反应同样具有出色的官能团耐受性,如-COOR(28、33、44、45),-CN(29、34),末端不饱和化学键(43、44)和杂芳烃(38-43、46)。一些含氟和氟代烷基的二芳基酮(30-32、36、37、39-42)也非常容易获得。此外,烷基卤化物也可实现亲电偶联反应,从而以高收率(高达92%)获得官能化的酮产物47-49。而使用苄基氯时,获得46%收率的49,但形成大量的副产物酯(可能由于亲核取代形成)。通过克级实验,同样获得82%收率的产物29。
![]()
为了进一步证明该反应的实用性,作者对一些生物活性的分子进行了相关的后期修饰(Fig. 6)。非诺贝特(Fenofibrate)是一种用于调节脂质水平和血液粘度的药物,可通过该反应一步制备获得65%收率的50。此外,一些复杂的溴代分子,也可通过上述方法合成相应的产物50-55。同时,该策略可以允许在早期引入官能团,避免后期引入所带来的各种弊端,如对收率、合成步骤、官能团的保护与脱保护等的不利影响。
紧接着,作者也进行了相关的对照试验(Fig. 7)。当Ar-Ni(II)中间体(56)与1.5当量Ph3P在DMF/MeCN混合溶剂中反应时,通过31P NMR分析未观察到配体交换。而在光氧化还原条件下,4-甲基苯甲酸与Ar-Ni(II)中间体(56)在标准条件下反应时,获得所需的C-C偶联产物(35),收率为42%,进一步证明了机理的正确性。
![]()
总结
南京大学谢劲课题组报道了通过光氧化还原、镍催化和正膦基自由基的协同作用实现芳基酸和芳基/烷基溴化物的亲电偶联反应,获得多种具有高度官能团化的酮衍生物。该反应具有底物廉价易得、官能团耐受性高(作为常规Weinreb酮合成的补充)、操作简单(体现了原子和步骤的经济性)等优点。此外, C-O键的断裂和随后的快速酰基自由基氧化加成速率,可控制选择性C-C键形成。

















No comments yet.