
本文作者:杉杉
导读
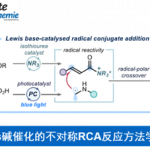
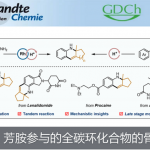
近日,上海有机化学研究所张国柱课题组在Angew. Chem. Int. Ed. 上发表论文,报道了通过直接C-H官能团化实现唑类化合物与1-芳基烷基溴化物的不对称烷基化反应。在蓝光激发下,铜(I)/咔唑基双恶唑啉(CbzBox)催化体系具有良好的反应性和高的立体选择性,从而可高效的构建手性烷基唑化合物。
Photo-Promoted Copper-Catalyzed Enantioselective Alkylation of Azoles
Chen Li, Bin Chen, Xiaodong Ma, Xueling Mo, and Guozhu Zhang
Angew. Chem. Int. Ed. ASAP DOI: 10.1002/anie.202009323 https://doi.org/10.1002/ange.202009323
正文
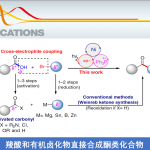
烷基化杂芳基化合物(alkylated heteroaromatics)作为一类重要的有机分子,具有独特的生物学、药学和材料学特性。Friedel-Crafts烷基化、Miniscitype自由基加成和金属催化的烯烃加氢杂芳基化是构建烷基化杂芳基化合物的传统方法。近年来,使用易得的碳卤化合物与杂环酸性C-H键的直接芳基化、烯基化和炔基化已取得重大地进展。但是,使用卤代烷进行直接烷基化更具挑战性,特别是含有β-氢的烷基化合物,易发生β-H消除、原脱卤和自偶联等副反应。因此,芳基杂环的不对称烷基化仍是一项具有难度的课题。
为了解决上述问题,近年来已开发出多种过渡金属催化的交叉偶联方法。基于导向基团的引入,能够允许杂芳基化合物的单个C-H键选择性烷基化。同时,杂芳基化合物酸性C-H键可实现直接烷基化。在过去的十年中,Miura[1a],Huang[1b],Zhou[2a]和Fu[2b]报道了在钯催化下实现杂芳基化合物与烷基卤化物之间的偶联反应。2012年,Hu[3]课题组首次报道了使用铜催化剂,实现唑类化合物与苄基溴的偶联(Scheme 1, eq 1)。2016年,Ohmiya[4a]课题组开发了一种铜催化体系,实现γ,γ-二取代伯烯丙基磷酸酯与唑类化合物的不对称烯丙基烷基化反应(Scheme 1, eq 2)。然而,对于铜催化苄基溴化物与唑类化合物的不对称烷基化反应尚未被报道[4b],相应的手性烷基唑类化合物通过文献中[5]的手性色谱柱拆分而获得。此外,不对称的C-H功能化具有原子和步骤经济性的特点,可轻松合成具有价值的光学活性分子。因此,迫切需要一种通过C-H功能化实现唑类化合物不对称烷基化的新策略。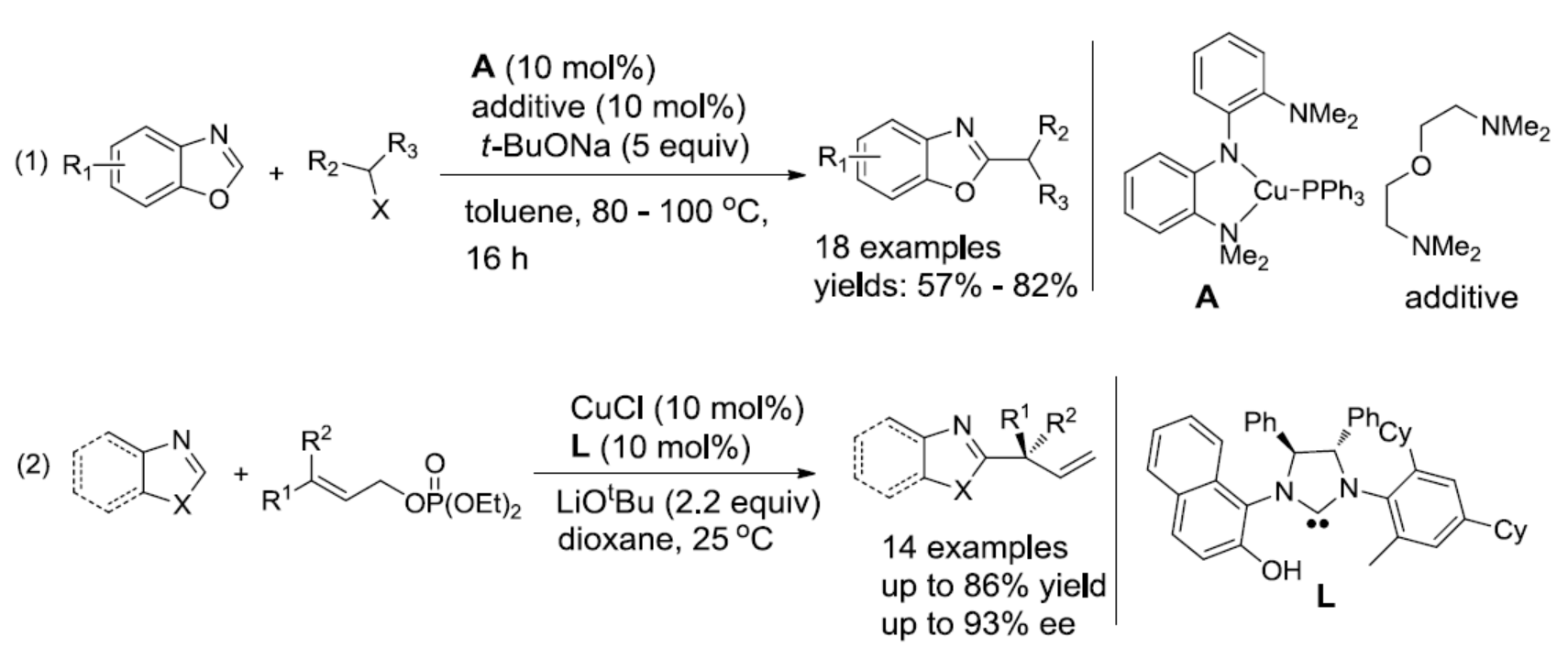
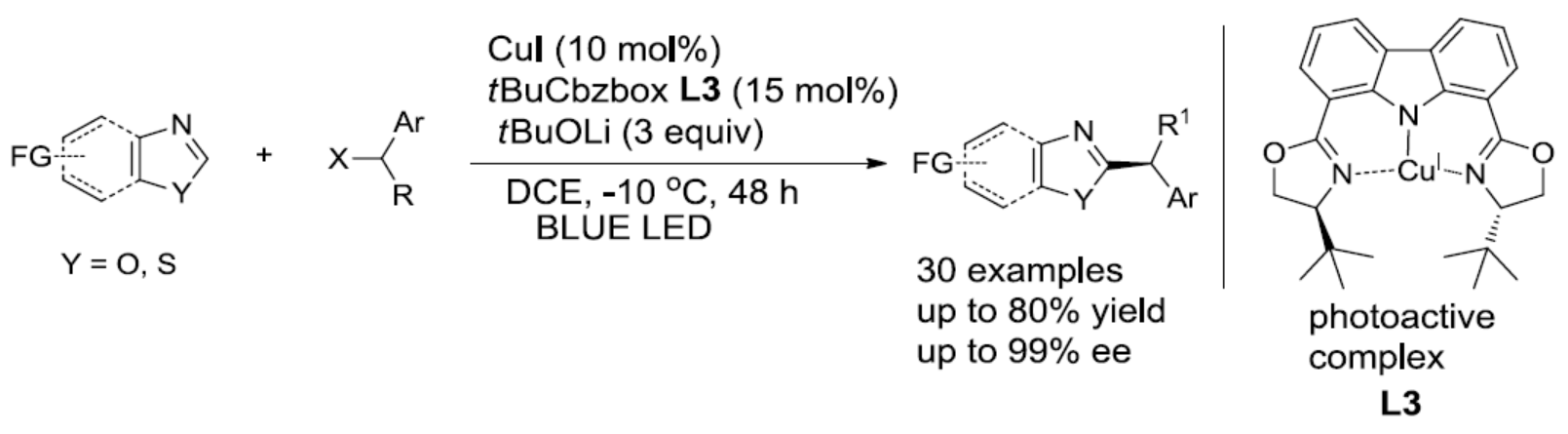
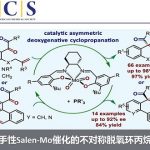
光氧化还原催化已成为一种强大的合成工具。Stephenson[6a-b]课题组报道了在室温的光催化条件下,实现吲哚、吡咯和呋喃与溴代丙二酸酯的烷基化反应。Fu[7a]课题组通过光诱导铜催化实现不对称C-N键的偶联。有机所张国柱课题组受上述结果的启发以及对铜催化的持续研究[8],报道了一种有效的光促进铜催化体系,实现唑类化合物的直接不对称烷基化反应。该反应涉及由仲苄基溴生成自由基的过程(Scheme 1, eq 3)。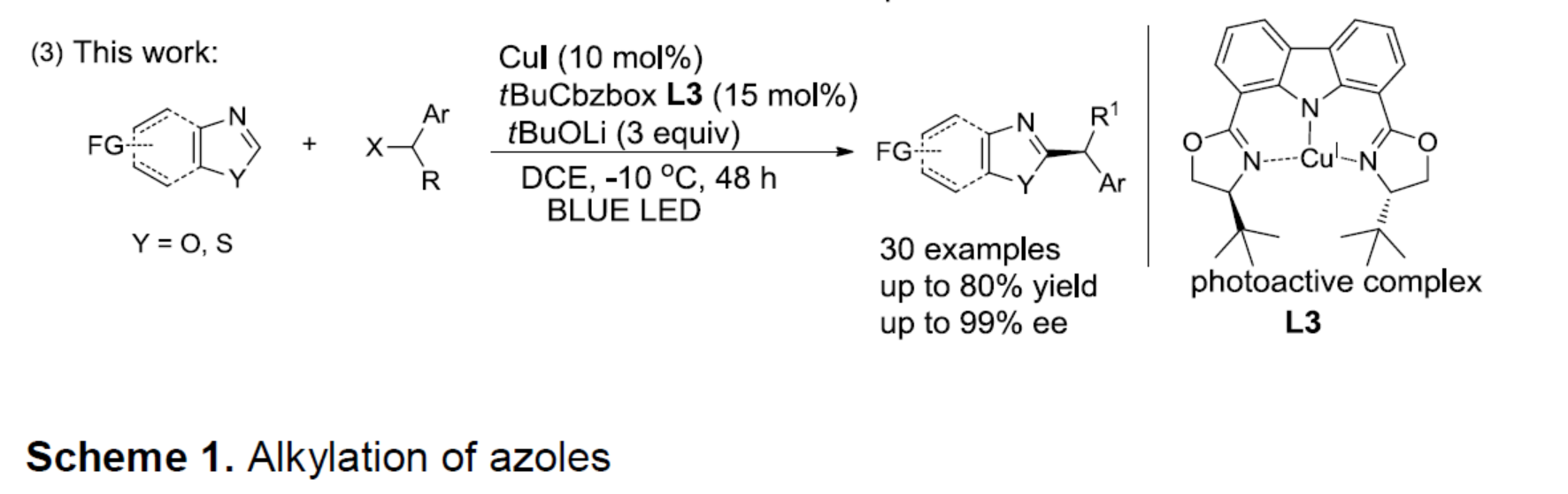
最近,该课题组报道了一种光诱导的铜催化体系,并使用二苯胺恶唑啉(BOPA)配体,实现烯烃的不对称三组分烷基/芳基炔基烷基化反应[8b]。作者推测,光激发的手性阴离子NNN-铜配合物可在温和的条件下通过单电子还原活化烷基溴来促进烷基自由基的生成,从而与杂芳烃结合形成碳-碳键[7b-d]。
首先,作者以苯并恶唑1a和(1-溴乙基)苯2a作为模型底物,进行了相关 反应条件的筛选(Table 1)。反应的最佳条件为,使用t-BuCbzBox L3作为手性配体,CuI作为催化剂,蓝色照射下于-10 ℃下反应48 h后,即可获得75%收率和90%ee的目标产物3a(entry 15)。
在获得上述最佳反应条件后,作者开始对苄基溴底物2进行了扩展(Table 2)。反应结果表明,具有不同长度的烷基链或含有无取代苯基的烷基链,均与体系兼容,获得相应的产物3b-3d。对位具有弱的给电子基(3e和3f),卤原子(溴、3g,氟,3h和3i),吸电子基团(如三氟甲基,3j和3k)、以及苯基(3l)的底物,均可获得相应的产物。同时,具有邻位取代的苄基溴化物(如Me和Cl),同样可获得相应的产物3p和3q。同样,含有萘(3m-3o)、杂环化合物(3r-3s)的底物,也与体系兼容。此外,烷基侧链的末端具有氯化物以及3-溴环己-1-烯底物,均可实现此类转化,从而获得相应的产物3t和3u。然而,当前条件不能获得相应的产物3v-3z。
随后,作者对苯并恶唑底物进行了相关的扩展(Table 3)。反应结果表明,具有给电子基(Me、MeO)以及吸电子基(F、COOMe)的底物,均可获得相应的产物4a-4d。萘并恶唑(4e和4f)、苯并噻唑(4h和4i),均实现此类转化,在和1-(1-溴戊基)萘反应时效果更好。同时,二取代底物(4j和4k)、和具有空间位阻的1-(1-溴戊基)萘(4l))反应时,同样取得较好的结果。此外,使用(1-溴乙基)苯为底物时,需使用CuTc作为铜源,才能获得相应的产物4m。然而,在标准条件下,未能获得芳基杂环产物4n-4p。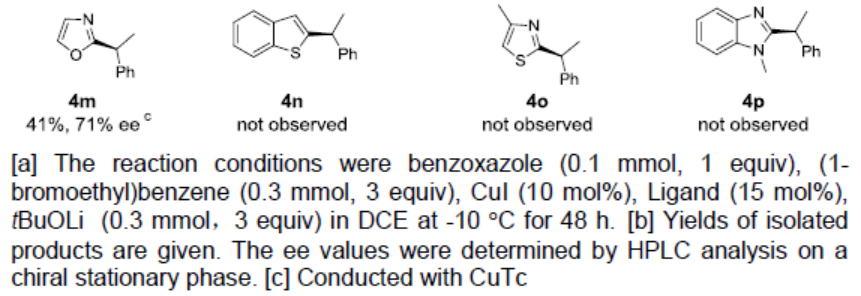
为了进一步了解反应的机理,作者进行了相关的氘代实验(Scheme 2)。首先,在室温下将1a与CuI和L3混合反应2小时,然后加入CD3OD(20当量),发现H/D交换(eq 1)。当1a与tBuOLi在室温反应2小时,然后加入CD3OD,1a的C2具有85%的氘,这表明恶唑C2的去质子化易发生(eq 2)。当将1a与tBuOLi,CuI和L3混合并在室温下搅拌2小时,然后加入CD3OD时,1a的H/D交换比约为88%,这表明铜配体配合物不影响锂化步骤。[Li+oxazole-]的金属转移最可能形成[LCu-oxazole]-Li+配合物(eq 3)。苄基氯与环丙基的反应仅提供开环产物,表明涉及自由基中间体(eq 4)。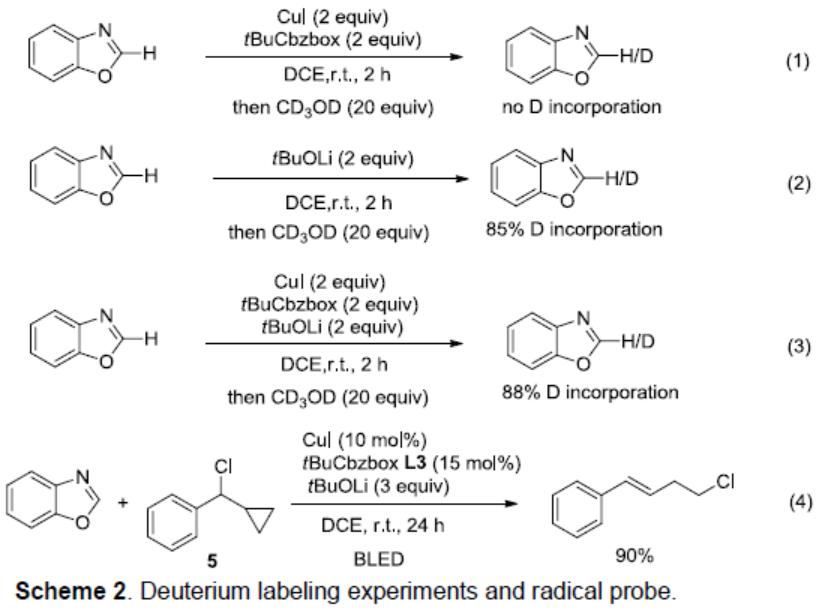
根据上述的实验和相关文献的查阅[9],作者提出了一种可能的反应机理(Figure 3)。在碱和配体存在下,原位形成[LCuI]配合物(A),然后与[Li-azole]进行金属转移生成[LCu-azole]-Li+中间体B。该中间体B作为光敏物质进行光激发,得到[LCu(I)-azole]-* Li+(C),后者将电子传递至烷基溴,生成LiBr和[LCuII-azole][R•](D)。该中间体通过对映选择性自由基捕获,迅速转变为手性Cu(III)中间体E。最终,从Cu(III)中心经还原消除形成光学纯苄基唑。此外,从D到产物的另一种直接SET过程也是可行的。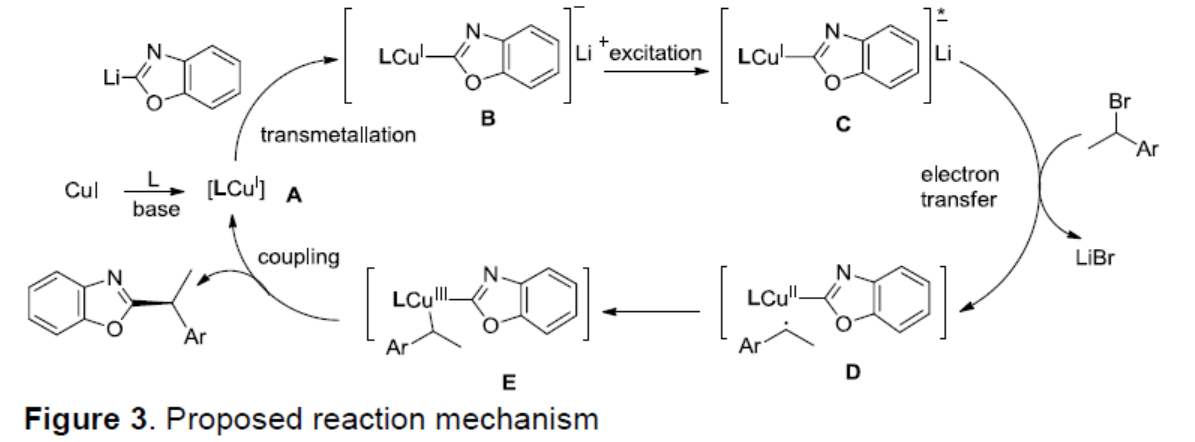
总结
上海有机所张国柱课题组报道了一种可见光促进铜催化体系,实现唑类化合物的不对称烷基化反应。该反应在低温下进行,并与各种唑类化合物相容。该体系成功的将可见光光氧化还原催化与铜催化的不对称C-H功能化相结合,并使用单一的铜CbzBox配合物同时作为光催化剂和手性催化剂。
参考文献
- [1] a) T. Yao, K. Hirano, T. Satoh, M. Miura, Chem. Eur. J. 2010, 16, 12307; b) P. Xie, H. Huang, Y. Xie, S. Guo, C. Xia, Adv. Synth. Catal. 2012, 354, 1692 –1700.
[2] a) X. J. Wu, J. W. T. See, K. Xu, H. Hirao, J. Roger, J. C. Hierso, J. R. Zhou, Angew. Chem. Int. Ed. 2014, 53, 13573 –13577; Angew. Chem. 2014, 126, 13791 –13795; b) B. Xiao, Z. J. Liu, L. Liu, Y. Fu, J. Am. Chem. Soc. 2013, 135, 616; c). X. Zhao, G. Wu, Y. Zhang, J. Wang, J. Am. Chem. Soc. 2011, 133, 3296-3299.
[3] P. Ren, I. Salihu, R. Scopelliti, X. Hu, Org. Lett. 2012, 14, 1748.
[4] a) H. Ohmiya, H. Zhang, S. Shibata, A. Harada, M. Sawamura, Angew. Chem., Int. Ed. 2016, 55, 4777-4780; Angew. Chem. 2016, 128, 4855-4858; b) during the revision of this manuscript, Liu reported a similar study: X.-L. Su, L. Ye, J.-J. Chen, X.-D. Liu, S.-P. Jiang, F.-L. Wang, L. Liu, C.-J. Yang, X.-Y. Chang, Z.-L. Li, Q.-S. Gu, X.-Y. Liu, Angew. Chem. Int. Ed. 2020, doi: 10.1002/anie.202009527.
[5] T. Kubota, N. Sawada, L. Zhou, C. J. Welch, Chirality, 2010, 22, 382-388
[6] a) L. Furst, B. S. Matsuura, J. M. R. Narayanam, J. W. Tuckerand, C. R. J. Stephenson, Org. Lett., 2010, 12, 3104; b) E. C. Swift, T. M. Williams, C. R. J. Stephenson, Synlett, 2016, 27, 754.
[7] a) Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters, G. C. Fu, Science 2016, 351, 681-684; b) S. E. Creutz, K. J. Lotito, G. C. Fu, J. C. Peters, Science, 2012, 338, 647-651; c) A. C. Bissember, R. J. Lundgren, S. E. Creutz, J. C. Peters, G. C. Fu, Angew. Chem., Int. Ed. 2013, 52, 5129-5133; Angew. Chem. 2013, 125, 5233-5237.
[8] a) Y. Xiong, X. Ma, G. Zhang, Org. Lett. 2019, 21, 1699−1703; b) Y. Zhang, Y. Sun, B. Chen, M. Xu, C. Li, D. Zhang, G. Zhang, Org. Lett. 2020, 22, 1490-1494; c) X. Mo, B. Chen, G. Zhang, Angew. Chem. Int. Ed. 2020, 59, 13998-14002; Angew. Chem. 2020, 132, 14102-14106.
[9] a) X. J. Wu, J. W. T. See, K. Xu, H. Hirao, J. Roger, J. C. Hierso, J. R. Zhou, Angew. Chem. Int. Ed. 2014, 53, 13573 –13577; Angew. Chem. 2014, 126, 13791 –13795; b) B. Xiao, Z. J. Liu, L. Liu, Y. Fu, J. Am. Chem. Soc. 2013, 135, 616; c) X. Zhao, G. Wu, Y. Zhang, J. Wang, J. Am. Chem. Soc. 2011, 133, 3296-3299. d).H. Q. Do, O. Daugulis, J. Am. Chem. Soc. 2007, 129, 12404 –12405.













No comments yet.