
本文作者:ChemBoy
导读
目前,已报道的烯丙基C-H键胺化反应,使用的胺化试剂均为酰胺、磺酰胺或碳酰胺,导致无法直接生成烷基烯丙基胺产物。最近,德国马普煤炭研究所Tobias Ritter教授课题组成功开发了一种基于噻蒽分子(Thianthrenes)的胺化试剂,在可见光促进的光催化反应条件下,可与烯烃反应直接生成烷基烯丙基胺化合物,其中的胺化试剂亚胺基噻蒽可通过一级胺一步制备。相关研究成果发表在《J.Am.Chem.Soc.》上:
“Allylic Amination of Alkenes with Iminothianthrenes to Afford Alkyl Allylamines”
Qiang Cheng, Junting Chen, Songyun Lin, and Tobias Ritter*
J.Am.Chem.Soc. 2020, 142, 17287. DOI: 10.1021/jacs.0c08248 https://doi.org/10.1021/jacs.0c08248
正文
前言
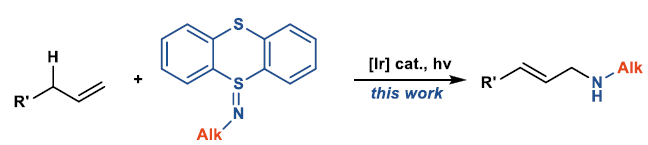
到目前为止,通过烯烃的烯丙基C-H键胺化可获得具有合成价值的烯丙基胺,如磺酰胺、碳酰胺、羧酰胺或N-杂芳环衍生物[1]。尤其在过渡金属催化中,通过形成π-烯丙基或氮宾类中间体,成为合成化学家构筑烯丙基胺衍生物的强大工具箱。虽然这些方法的合成应用价值无可争议,但所用的胺基化试剂具有较大的局限性,需要在氮原子上引入合适的取代基以降低氮原子上的电子云密度,常见的是酰胺(磺酰胺)或碳酰胺[2]。在这里,Ritter课题组报道了首例烯丙基C-H键胺化反应,直接合成烷基烯丙基胺化合物,其中以噻蒽亚胺作为胺化试剂,此胺化试剂可通过一级胺经一步简单反应制备,这种胺化试剂与烯烃在合适的光敏剂存在、可见光辐射的条件下,即可顺利发生烯丙基C-H键胺化反应。作者认为,在该反应中C-N键的形成经历了光敏剂与胺化试剂噻蒽亚胺在光辐射下发生能量转移生成的氮中心自由基(NCRs)与烯烃加成的过程。该过程在概念上与其他已知的烯烃与亚氨基噻蒽的胺化反应不同。端烯与内烯均能顺利发生转化。这种不同寻常的方法为合成通过其他的C-H键官能团化反应无法直接获得的目标烯丙基胺衍生物提供一种新途径。
烯丙基胺可通过SN2或是其他烯丙基烷基化反应来合成,如Tsuji-Trost反应[3]、Overman重排[4]、亚胺的烯基化[5]、烯酮或烯醛的还原胺化[6]以及联烯或二烯的氢胺化反应[7]。而由烯丙基C-H键直接转化为C-N键的反应方法稀缺。目前,烯丙基C-H键直接胺化的反应策略主要有两种:一种是在过渡金属催化下,通过氮亲核试剂进攻反应过程中形成的π-烯丙基金属中间体,生成C-N键(Scheme 1A)[8],这种策略仅适用于磺酰胺、碳酰胺或N-杂芳烃类含氮亲核试剂,而烷基胺类亲核试剂在该类方法中的反应活性较低;另一种是通过C-H键插入金属氮宾物种从而实现烯丙基C-H键胺化(Scheme 1B)[9],而这种策略仅局限于磺酰胺和碳酰胺类胺化试剂,由于这类胺试剂可与过渡金属形成金属氮宾类物种并且不会发生Curtius重排。而烷基胺不常用于金属氮宾物种介导的C-H键胺化反应,可能是由于形成的金属氮宾物种中存在很强的金属-配体多重键,导致与C-H键发生反应的活性降低[10]。此外,在很多过渡金属催化的烯烃与氮宾的转移反应过程中,通常会出现与烯丙基C-H胺化反应的竞争反应,即吖啶化反应(Scheme 1B)[11]。总之,目前已经报道了的分子间烯丙基胺化反应中所使用的胺化试剂均为酰胺、磺酰胺或碳酰胺类氮亲核试剂(Scheme 1C)。Ritter研究团队在这里报道了一种利用基于噻蒽的胺化试剂,实现与烯烃的烯丙基C-H键的直接胺化反应,生成烷基烯丙基胺化合物(Scheme 1D)。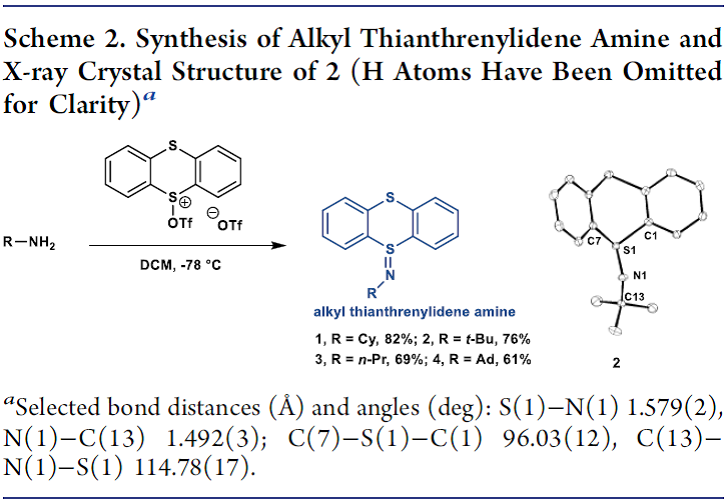
条件优化
该研究团队在之前就已经报道了一种一锅法合成烷基噻蒽亚胺类胺化试剂的方法,一级烷基胺与噻蒽硫氧化物在三氟乙酸酐的活化作用下迅速反应生成亚胺基噻蒽(Scheme 2)。无论是叔丁基胺还是线型烷基亚胺基噻蒽均能通过该方法获得(1-4),并且这种亚胺对空气、湿气及硅胶柱均不敏感,能够在室温条件下空气氛围中稳定保存至少一年。
首先对反应条件进行筛选优化,作者发现以4-甲基戊-1-烯作为底物、环己基亚胺基噻蒽1作为胺化试剂、铱配合物Ir[dF(CF3)ppy]2(dtbpy)PF6(E°(IrIII*/IrII) = 1.21 V vs SCE and E°(IrIII*/IrIV) = −0.89 V vs SCE),)作为光敏剂,在以HFIP作为反应溶剂、TFAA存在、反应温度8 °C、可见光照射条件下反应能够以71%的产率获得目标产物烷基烯丙基胺11(E:Z=16:1)(Scheme 3)。在该反应中,光与光敏剂是必须得。还原性更强的光敏剂Ir(ppy)3 (E°(IrIII*/IrIV) = -1.73 V vs SCE)或氧化性更强的光敏剂Mes-Acr+ (E°(M*/M−)= 2.15 V vs SCE)在该反应中均不能获得目标胺化产物。该反应中所使用的光催化剂会使烷基胺被氧化,酸性溶剂HFIP与三氟乙酸酐的组合能够有效抑制该副反应的发生,从而提高目标产物的产率。作者发现TFAA在该反应中不是起酰基化试剂的作用,认为是与质子性溶剂HFIP反应提供质子酸同时起到吸水剂的作用。非质子性溶剂如乙腈不能获得目标烯丙基,并且低温条件下能够抑制胺化试剂N-S键断裂生成一级胺的副反应发生。作者还发现过量的胺化试剂会引起反应产率降低,这可能是因为过量的亚氨基噻蒽会导致体系变成深紫外吸收溶液会阻止蓝色可见光的吸收。
底物拓展
在确定最优反应条件后,作者将蓝光灯换成紫光灯对反应的底物范围进行了研究(Scheme 4)。胺化试剂1与末端烯烃反应能选择性的获得反式-烷基烯丙基胺产物(6-17, 30)。作者观察到该反应更喜欢化学选择性的发生烯丙基胺化,当底物存在苄基C-H键时选择性的发生烯丙基C-H键胺化反应(6, 20),以及烯丙位存在支链时能够提高反式选择性(9-11)。,烯丙基溴化物13的成功合成证明该反应条件温和,化合物13易发生分子内SN2’取代反应生成吖啶。该反应的官能团兼容性较好,酰亚胺(7)、酯(8,26,30)、硅基醚(12)、醇(14, 22, 28),羧酸(15)以及酮(30)均能兼容;此外,其他不饱和官能团如芳烃(6, 20)、炔烃(16)也能兼容。对于含有多个双键的烯烃底物,只观察到单一胺化产物(17, 27, 29),这类底物反应会选择性地在更富电子的烯烃上发生(26, 28)。三取代的烯烃也能够顺利发生反应生成目标烯丙基胺(18, 19)。作者发现底物内烯的双键构型与产物烯丙基胺的双键构型无必然联系,顺式-和反式-4-辛烯底物反应后生成的产物构型相同(21);两个不同取代基取代的1,2-二取代烯烃(24)的区域选择性会比相同取代基取代的1,2-二取代烯烃烯烃(25)的区域选择性高。当使用烯丙基芳烃作为底物时,所生成的烯丙基胺产物以顺式构型为主(20, 24),而其他脂肪取代基类烯烃底物所生成的产物以反式构型为主。此外,作者发现共轭二烯、烯丙基醇和烯丙基胺类底物在该反应条件下不适用。紧接着,作者还对噻蒽类胺化试剂的范围进行了评估。大位阻与线型的烷基胺化试剂(1-4)在这种烯丙基的C-H键胺化反应中均能够顺利进行(31-35);氨基醇也能兼容(37),而未保护的氨基酸不能用于合成相应的亚胺基噻蒽。
机理研究
最后,作者对反应机理进行了研究(Scheme 5)。作者通过荧光猝灭实验、循环伏安法、其他能量转移催化剂的分析、UV辐射以及计算实验的结果提出在该反应体系中,光催化剂不是作为光氧化还原催化剂,而是作为能量转移催化剂。Stern-Volmer荧光猝灭实验结果表明激发态的铱光催化剂被质子化的亚胺噻蒽有效猝灭(Scheme 5a);除了动态猝灭外,基态猝灭剂与光催化剂发生预络合能够引起静态猝灭,从而可以合理解释所观察到的非线性关系。作者测定了质子化的1(A)的氧化还原电势(Eox = 2.0 V vs Ag/AgCl, Ered = −1.4 V vs Ag/AgCl),发现与光催化剂的氧化还原电势和反应的产率无相关性。作者发现该胺化反应活性与光催化剂的第一三线激发态的能量高低相关(Scheme 5b), 反应中所用的光催化剂的三重激发态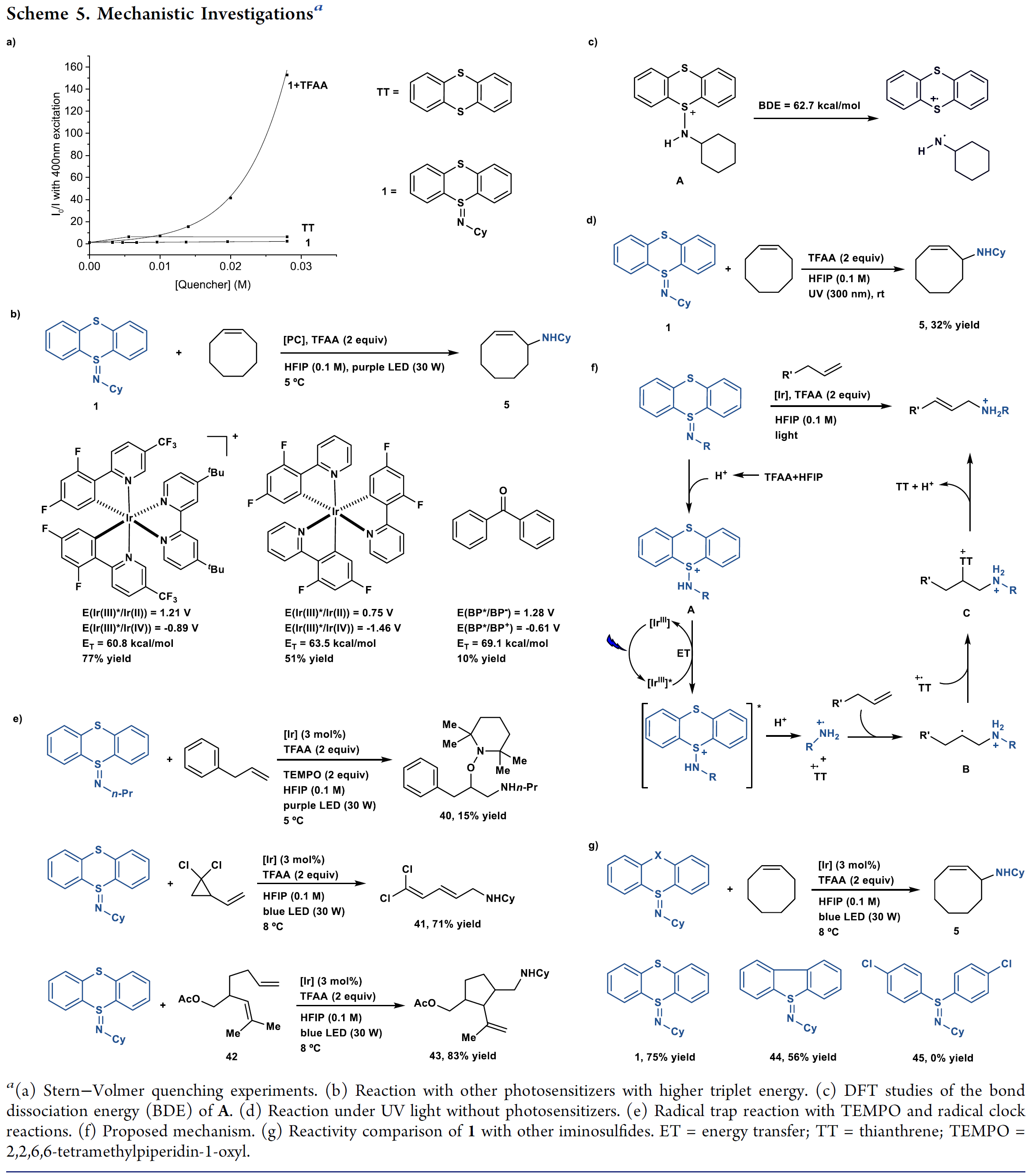
能量为61 Kcal/mol,并且寿命长(2.3 μs)。作者还尝试对A的第一激发三重态的能量进行了计算(Scheme 5c),观察到N-S键的断裂无能垒,这与自由基的形成一致,但无法从A获得的三重态能量到铱催化剂的三重态能量的比较。取而代之,作者计算出A中S键的基态键解离能是63 Kcal/mol,这说明A中N-S键比较弱,在合适的光敏剂作用下就能发生均裂。为了支撑A中的N-S键弱这一观点,作者发现在没有光敏剂存在时,反应在UV辐射下也能够获得目标胺化产物(Scheme 5d)。所有的数据结果都与该反应中的光敏剂是能量转移催化剂的作用这一观点保持一致,作者提出了可能的催化循环过程(Scheme 5f)。氮中心自由基更喜欢提取氢原子而不是与π-体系发生加成,通过质子化提高氮中心的亲电性使其更喜欢发生加成反应。在该反应体系中,氮中心自由基与质子性溶剂形成强氢键导致其带部分正电荷,接着与烯烃发生区域选择性的加成生成自由基B。碳自由基B可能与噻蒽自由基阳离子很快发生自由基偶联反应形成C,最后C发生消除或碳正离子直接去质子化生成产物烯基胺。作者通过自由基捕获实验与自由基钟实验进一步证明了该反应的机理(Scheme 5e)作者认为噻蒽自由基阳离子的持久性对该反应的反应活性至关重要,这促使直接生成烷基烯丙基胺。作者发现其他类亚胺基硫化物不能很好甚至不能发生反应(Scheme 5g)。
总结与评价
Ritter教授课题组开发了一种新型光催化烷基胺与烯烃的烯丙基C-H胺化反应的方法。该方法能够实现过渡金属催化烯丙基C-H活化或氮宾转移反应不能实现的烯丙基胺化反应,同时可一步合成烷基烯丙基胺化合物,从而打破了吸电子基取代胺化试剂的局限。此外,该反应具有良好的官能团耐受性、广泛的底物适范围等优点。
(注:本文中图片均来自J. Am. Chem. Soc. 2020, 142, 17287)
参考文献
- [1] Ramirez, T. A.; Zhao, B.; Shi, Y. Chem. Soc. Rev. 2012, 41, 931.DOI:10.1039/c1cs15104e
[2]Trowbridge, A.; Walton, S. M.; Gaunt, M. J. Chem. Rev.2020, 120, 2613.DOI:10.1021/acs.chemrev.9b00462
[3] Trost, B. M.; van Vranken, D. L. Chem. Rev.1996, 96, 395. DOI: 10.1021/cr9409804
[4] Overman, L. E.; Carpenter, N. E. Org. React.2005, 66, 1.DOI:10.1002/0471264180.or066.01
[5] Brak, K.; Ellman, J. A. J. Am. Chem. Soc.2009, 131, 3850.DOI: 10.1021/ja9002603
[6] Abdel-Magid, A. F.; Carson, K. G.; Harris, B. D.; Maryanoff, C. A.; Shah, R. D. J. Org. Chem.1996, 61, 3849. DOI: 10.1021/jo960057x
[7] Huang, L.; Arndt, M.; GooBen, K.; Heydt, H.; Gooben, L. J. Chem. Rev.2015, 115, 2596. DOI: 10.1021/cr300389u
[8] Lei, H.; Rovis, T. J. Am. Chem. Soc.2019, 141, 2268. DOI: 10.1021/jacs.9b00237
[9] Shin, K.; Kim, H.; Chang, S. Acc. Chem. Res.2015, 48, 1040.DOI: 10.1021/acs.accounts.5b00020
[10]Baek, Y.; Hennessy, E. T.; Betley, T. A. J. Am. Chem. Soc.2019, 141, 16944.DOI: 10.1021/jacs.9b09015 - [11] Alderson, J. M.; Corbin, J. R.; Schomaker, J. M. Acc. Chem. Res.2017, 50, 2147.DOI: 10.1021/acs.accounts.7b00178















No comments yet.