
本文作者:杉杉
导读
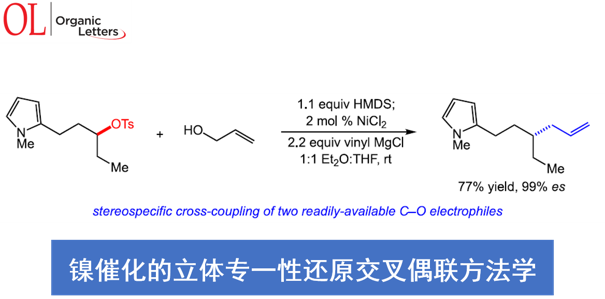
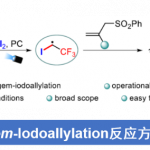
采用廉价易得的亲电底物参与的立体专一性交叉偶联反应方法学,在实现C-C键构建的相关研究中具有重要意义。近日,North Carolina大学的E. J. Alexanian课题组在Org. Lett.中发表论文,报道一种在镍催化条件下,烯丙醇与手性、非外消旋的烷基对甲苯磺酸酯之间的还原交叉偶联反应方法学。进而能够高度立体专一性地获得一系列具有合成应用价值的烯丙基砌块。并且,这一全新的催化体系由简单的镍盐与商品化的烯丙基Grignard试剂构成。同时,在这一全新的C-C键构建策略中,涉及两种具有C-O键的亲电底物之间的交叉偶联反应过程。
Stereospecific Nickel-Catalyzed Reductive Cross-Coupling of Alkyl Tosylate and Allyl Alcohol Electrophiles
Q. D. Tercenio, E. J. Alexanian, Org. Lett. 2021, 23, 7215. doi: 10.1021/acs.orglett.1c02616.
正文
通过简单的手性、非外消旋亲电底物参与的催化立体专一性C-C键形成反应方法学在有机合成领域的相关研究中,具有极为重要的意义。同时,这一策略能够有效地应用于一系列对映选择性合成方法学的相关研究。近期,对于这一全新的合成转化策略的相关研究,已经取得较大进展。并且,通过这一策略,能够成功将一系列对映富集的苄基或脂肪族二级醇底物应用于采用多种不同金属催化剂参与的合成转化方法学研究[1]。例如,本课题组前期已经报道通过钴酸盐 (cobaltate)催化剂参与的烷基对甲苯磺酸酯的立体专一性羰基化反应方法学,进而顺利完成一系列羰基化合物的简洁不对称合成[2]。同时,近期,本课题组开始关注于通过二级烷基对甲苯磺酸酯参与的立体专一性亲电交叉偶联反应方法学中,底物应用范围的相关研究。
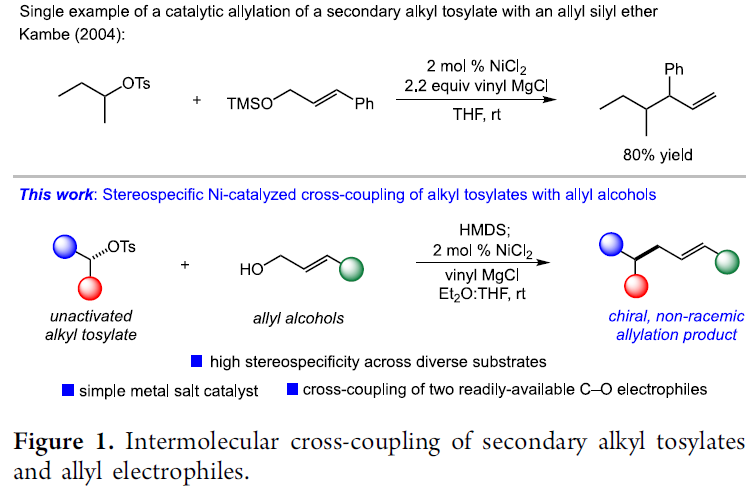
通过各类有机亲电底物参与的立体选择性烯丙基化反应方法学,是当代有机合成化学中实现C-C键构建的一种极为有效的反应策略。而且,本课题组同样已经开始关注于采用非活化的二级烷基对甲苯磺酸酯参与的立体专一性烯丙基化方法学的相关研究。尤为重要的是,本课题组已经报道采用各类简单的C-O烯丙基亲电分子作为偶联参与物,成功设计出一系列的全新的交叉偶联反应策略,进而能够有效避免具有较高反应活性的有机金属试剂的原位制备。并且,在立体专一性烷基-烯丙基偶联反应方法学的相关报道中,最具代表性的则是Jarvo课题组发展的通过2-乙烯基-4-卤代四氢吡喃底物参与的分子内环化反应方法学,进而顺利获得一系列乙烯基环丙烷衍生物[3]。而且,近期,同样有文献报道采用钴催化剂以及镍催化剂促进的烷基溴与烯丙基乙酸酯或碳酸酯之间的立体消失性还原烯丙基化 (stereoablative reductive allylation)反应方法学[4],其反应过程中涉及自由基中间体。
接下来,本课题组开始关注于在镍催化条件下,将两种C-O亲电底物应用于立体专一性烷基-烯丙基交叉偶联策略设计的可行性的研究。其中,较为有趣的是,在镍催化的烯丙醚与氯硅烷之间的硅基化策略的相关研究中,Kambe小组通过对于烷基对甲苯磺酸酯与烯丙醚之间偶联过程的深入研究,进而为上述烷基-烯丙基交叉偶联策略可行性的后续研究提供初步的依据[5]。然而,Kambe小组的相关报道并未涉及反应过程的立体选择性控制 (Figure 1)。同时,对于手性、非外消旋烷基对甲苯磺酸酯与简单烯丙基C-O亲电底物 (尤其烯丙醇)之间的立体专一性偶联反应方法学的成功设计,能够有效地实现两种廉价易得的偶联参与物之间的交叉偶联过程,进而显著提升这一偶联策略在有机合成中的应用潜力。受到上述相关报道的启发,这里,Alexanian课题组成功设计出通过两种C-O亲电底物参与的立体专一性还原交叉偶联反应方法学,进而成功实现相应C-C键的构建。
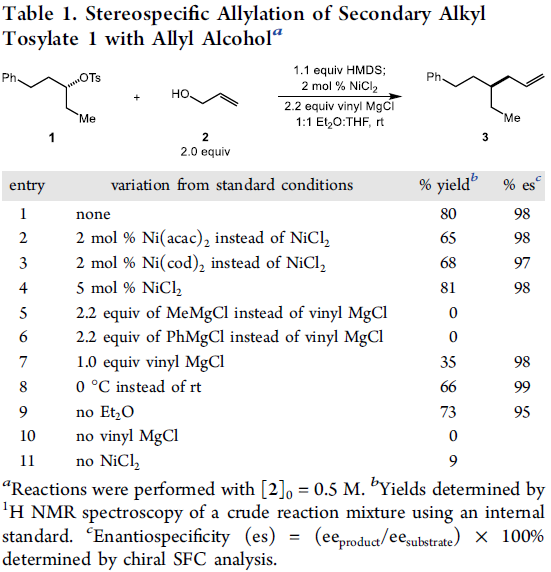
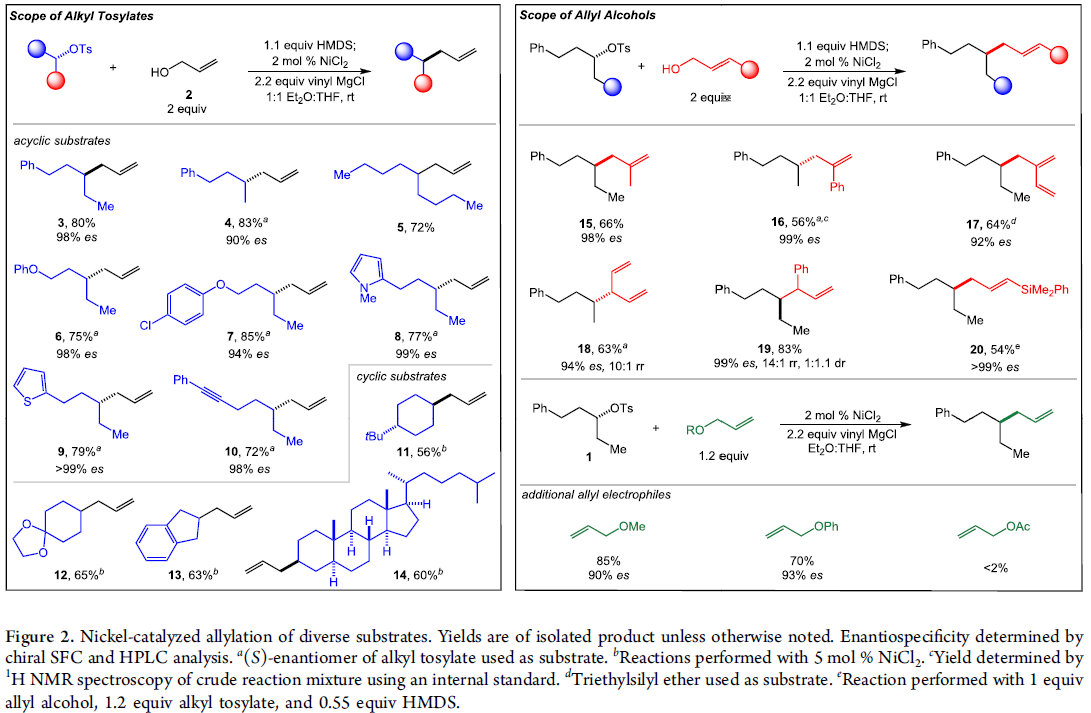
首先,作者采用对甲苯磺酸二级酯1与烯丙醇2作为模型底物,进行相关偶联反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:在采用HMDS对醇底物进行原位硅基化之后,再采用NiCl2作为催化剂,乙烯基氯化镁作为还原剂,在Et2O/THF (1:1 v/v)混合溶剂中,反应温度为室温,并以良好的反应收率与优良的立体专一性,获得相应的手性烯丙基化产物3 (80% 收率, 98% es)。
在上述的最佳反应条件下,作者首先对各类烷基对甲苯磺酸酯底物的应用范围进行考察 (Figure 2)。研究表明,一系列不同类型的二级烷基对甲苯磺酸酯 底物 (3–5),均能够有效地参与上述的还原偶联过程。之后,作者发现,上述的标准反应条件同样能够良好地与二级烷基对甲苯磺酸酯底物中存在的芳基醚基团 (6)、氯代芳基基团 (7)、芳香杂环基团 (8与9)以及炔基基团 (10) 进行兼容,并获得优良的化学选择性与对映专一性。同时,该小组进一步发现,各类具有环状结构的二级对甲苯磺酸酯,同样能够有效地参与上述的还原偶联过程,并获得相应的手性产物11–14。
之后,该小组对各类烯丙醇底物的应用范围进行深入研究。实验发现,一系列2-取代的烯丙醇底物,均能够以良好的反应收率与优良的立体专一性,获得相应的手性产物15–17。同时,研究表明,上述的标准反应条件对于戊-1,4-二烯-3-醇底物,同样能够以高度的区域选择性,获得相应的手性产物18。并且,该小组发现,上述的标准反应体系对于1,2-二取代烯基底物,同样能够以良好的反应收率与优良的对映专一性,获得相应的手性产物19。然而,由于支链产物的形成,因此,无法实现优良的非对映选择性控制。接下来,作者发现,通过1,2-二取代烯基底物参与的上述还原偶联过程,更利于形成线性产物,并表现出优良的立体选择性控制。此外,该小组进一步发现,其它不同类型的烯丙醚,例如甲基烯丙醚以及苯基烯丙醚,同样能够良好地参与上述的还原交叉偶联过程。然而,在过渡金属催化的烯丙基化反应方法学研究中最为常用的乙酸烯丙酯底物,则无法有效地参与上述的还原交叉偶联反应。
同时,作者对这一全新的还原偶联策略的合成实用性进行进一步研究 (Scheme 1)。该小组发现,通过上述的还原交叉偶联策略,能够成功实现Alpha-2B受体激动剂 (alpha-2B receptor agonist)24的不对称合成。其中,采用手性、非外消旋的对甲苯磺酸酯21作为起始原料,在上述的最佳反应条件下,与烯丙醇偶联参与物进行相应的立体专一性烯丙基化过程,进而能够以优良的立体选择性,获得关键砌块22。之后,通过22的闭环复分解过程,以良好的反应收率,获得相应的环己烯砌块23。接下来,通过后续的硅基去保护、氧化以及Van Leusen咪唑合成的三步反应过程,最终完成Alpha-2B受体激动剂分子24的构建。

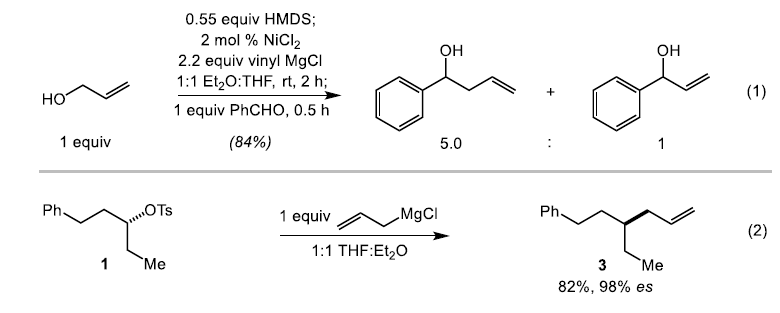
接下来,作者针对上述反应过程中,相关亲核试剂的差异,提出两种可能的反应机理: (1) 通过催化过程,形成相应的烯丙基Grignard试剂或 (2) 通过催化过程,形成相应的亲核性烯丙基镍酸盐 (allyl nickelate)配合物。之后,作者进行相关的控制实验研究。首先,该小组将原位形成的烯丙基硅醚在上述的标准条件下进行反应,之后,通过苯甲醛进行淬灭 (eq 1)。这里,作者假设,如果反应过程中能够形成相应的烯丙基Grignard试剂,则能够获得预期的烯丙基化产物。研究表明,上述的还原交叉偶联反应过程,能够以良好的反应收率,分别获得相应的烯丙基化与乙烯基化产物的混合物 (产物比为5.0:1),这一实验观察与烯丙基Grignard试剂的形成相一致。此外,作者发现,将烯丙基氯化镁试剂加入至对甲苯磺酸酯(S)-1时,同样能够以良好的反应收率与优良的立体专一性,获得相应的偶联产物3。这一实验事实同样符合烯丙基Grignard试剂作为上述交叉偶联过程中活性亲核试剂的设想 (eq 2)。
基于上述的实验研究,作者提出一种合理的反应机理 (Scheme 2)。首先,通过镍催化剂前体的还原过程,形成镍(0)丁二烯配合物,并进一步与通过烯丙醇原位形成的烯丙基硅醚分子经历相关的氧化加成过程,获得烯丙基镍配合物。之后,通过烯丙基镍配合物与乙烯基Grignard试剂之间的转金属化步骤,形成相应的烯丙基镍(II)乙烯基配合物,继而经历后续的亲核进攻过程,形成烯丙基镍酸盐 (allyl nickelate)配合物。接下来,通过烯丙基镍酸盐配合物的转金属化过程,产生相应的烯丙基Grignard亲核试剂,进而与烷基对甲苯磺酸酯底物作用,最终高度的立体专一性地获得相应的还原偶联产物。

总结
North Carolina大学E. J. Alexanian课题组成功设计出一种烷基对甲苯磺酸酯与烯丙醇之间的立体专一性还原交叉偶联反应方法学,进而获得一系列相应的手性烯丙基化产物。这一全新的还原交叉偶联策略中,采用简单的催化体系以及两种廉价易得的C-O亲电底物,进而有效地实现相应C-C键的构建。同时,反应机理研究表明,上述的偶联过程中,涉及烯丙基Grignard试剂的形成。
参考文献
[1] (a) E. J. Tollefson, L. E. Hanna, E. R. Jarvo, Acc. Chem. Res. 2015, 48, 2344. doi: 10.1021/acs.accounts.5b00223.(b) L. Cheng, N. P. Mankad, Chem. Soc. Rev. 2020, 49, 8036. doi: 10.1039/D0CS00316F.
(c) J. Xu, O. P. Bercher, M. R. Talley, M. P. Watson, ACS Catal. 2021, 11, 1604. doi: 10.1021/acscatal.0c05484.
(d) C. Yang, Z. Zhang, J. Liang, J. Liu, X. Lu, H. Chen, L. Liu, J. Am. Chem. Soc. 2012, 134, 11124. doi: 10.1021/ja304848n.
[2] (a) B. T. Sargent, E. J. Alexanian, J. Am. Chem. Soc. 2017, 139, 12438. doi: 10.1021/jacs.7b07983.(b) B. T. Sargent, E. J. Alexanian, Angew. Chem. Int. Ed. 2019, 58, 9533. doi: 10.1002/anie.201905173.
(c) H. Shenouda, E. J. Alexanian, Org. Lett. 2019, 21, 9268. doi: 10.1021/acs.orglett.9b03706.
[3] L. W. Erickson, E. L. Lucas, E. J. Tollefson, E. R. Jarvo, J. Am. Chem. Soc. 2016, 138, 14006. doi: 10.1021/jacs.6b07567. [4] (a) X. Qian, A. Auffrant, A. Felouat, C. Gosmini, Angew. Chem. Int. Ed. 2011, 50, 10402. doi: 10.1002/anie.201104390.(b) Y. Dai, F. Wu, Z. Zang, H. You, H. Gong, Chem. – Eur. J. 2012, 18, 808. doi: 10.1002/chem.201102984.
(c) L. L. Anka-Lufford, M. R. Prinsell, D. J. Weix, J. Org. Chem. 2012, 77, 9989. doi: 10.1021/jo302086g.
[5] (a) J. Terao, H. Watabe, H. Watanabe, N. Kambe, Adv. Synth. Catal. 2004, 346, 1674. doi: 10.1002/adsc.200404192.(b) J. Terao, H. Watanabe, A. Ikumi, H. Kuniyasu, N. Kambe, J. Am. Chem. Soc. 2002, 124, 4222. doi: 10.1021/ja025828v.
(c) J. Terao, A. Ikumi, H. Kuniyasu, N. Kambe, J. Am. Chem. Soc. 2003, 125, 5646. doi: 10.1021/ja034201p.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载













No comments yet.