
本文作者:杉杉
导读
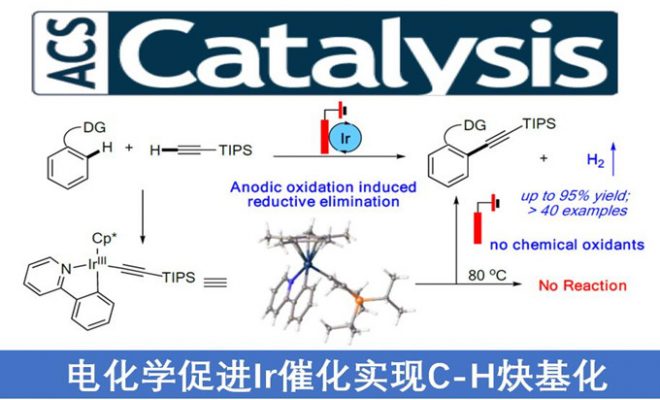
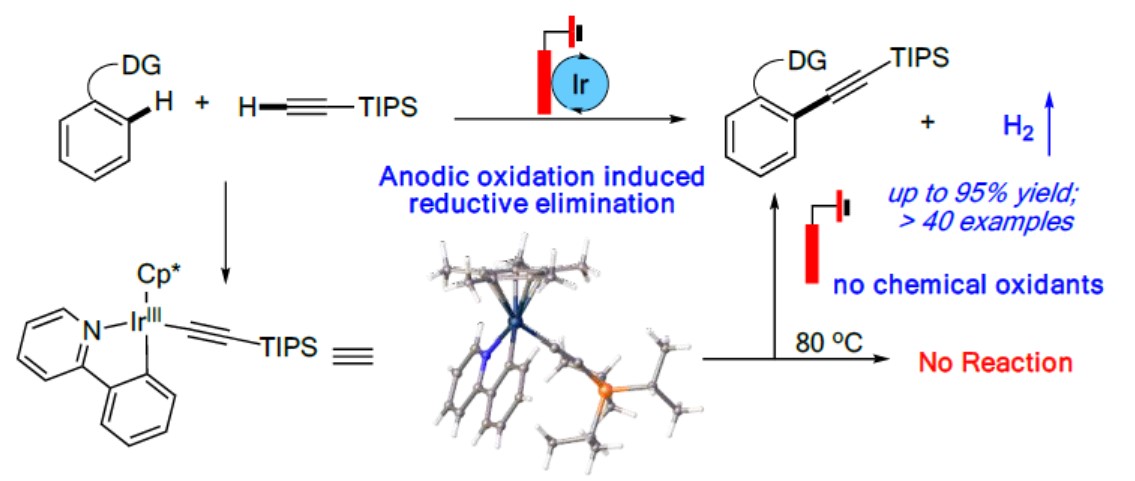
近日,南佛罗里达大学史晓东和衢州学院谢艳课题组共同在ACS Catal.上发表论文,报道了一种通过电化学促进铱催化的末端炔烃的直接C-H炔基化反应的方法。该反应使用Ir(III)中间体的阳极氧化来促进还原消除,从而以优秀的收率(高达95%)获得所需的偶联产物,避免了外部氧化剂的使用。同时,该反应仅产生H2作为唯一副产物,从而体现了高原子经济性。
Facilitating Ir-Catalyzed C-H Alkynylation with Electrochemistry: Anodic Oxidation Induced Reductive Elimination
Xiaohan Ye, Chenhuan Wang, Shuyao Zhang, Jingwen Wei, Chuan Shan, Lukasz Wojtas, Yan Xie, and Xiaodong Shi
ACS Catal. 2020, 10, 11693−11699 DOI: 10.1021/acscatal.0c03207
正文
在过去的二十年中,过渡金属催化的C-H功能化迅速发展,作为直接构建C-C键的高效方法。在已报道的文献中,导向基团的策略可实现位置选择性的引入新官能团 [1],而金属催化的C-H炔基化是引入炔基官能团的有效策略[2]。目前,促进氧化C-H炔基化存在两种主要的方法,(1)使用外部化学氧化剂(path a)(如Cu(OAc)2,AgOAc等),(2)采用氧化还原活性炔烃作为反应物和氧化剂(path b)(如乙炔基苯并恶唑酮试剂(EBX)或炔基溴化物)。尽管已取得很大的进展,但在有机合成中的实际性仍存在局限。因此,迫切需要一种可替代方法以高效的实现与末端炔烃的C-H炔基化。
在过去的几年中,电化学阳极氧化已成为化学合成中一种具有吸引力的策略。阳极氧化的优势在于可控的电池电势(Ecell),无需外部氧化剂即可将过渡金属阳离子氧化成较高的氧化态。尽管在过去的几年中已取得巨大的进步,但是对于某些转化而言,实现温和的条件和良好的官能团兼容性仍然是一项挑战。如炔烃可以通过迁移插入而与相应的金属环发生反应,从而在电化学条件下生成环化产物[3-4](path c)。在此,南佛罗里达大学史晓东和衢州学院谢艳课题组共同报道了通过电化学促进铱催化的C-H炔基化,通过阳极氧化促进还原消除(Scheme 1B)。该策略允许在温和条件下实现末端炔烃(无外部化学氧化剂)的直接C-H炔基化。
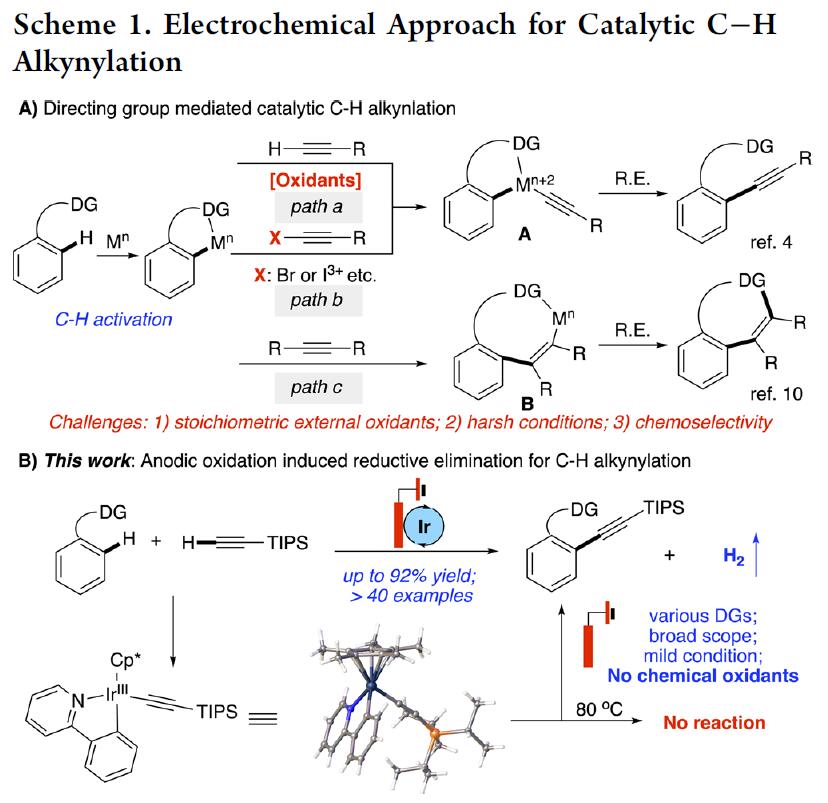
Li课题组报道了通过Rh(III)和Ir(III)催化实现EBX(作为反应物和氧化剂)的C-H炔基化反应,但这种转化的原子经济性很差[5]。最近,Xu课题组报道了Rh(III)配合物可快速活化C-H键,并将其应用于电化学氧化条件下直接构筑C-P键[6](Figure 1A)。受此启发,是否这种化学方法可扩展到具有挑战性的C-H炔基化反应中,从而避免了潜在的竞争性炔烃环化反应。
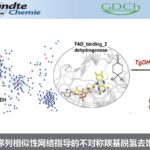
首先在有或无炔烃时,进行1a与Rh(III)或Ir(III)配合物之间的反应。空气稳定的Cp*Ir(III)配合物2a是通过1a直接C-H活化获得,配合物2b是通过往2a中加入炔烃获得(Figure 1B)。值得注意的是,C-H活化和炔烃加成过程均在室温下完成,突出了Ir(III)配合物在促进这些转化中的效率。同时,该反应是通过定向C-H活化获得铱-乙炔配合物的第一个例子。
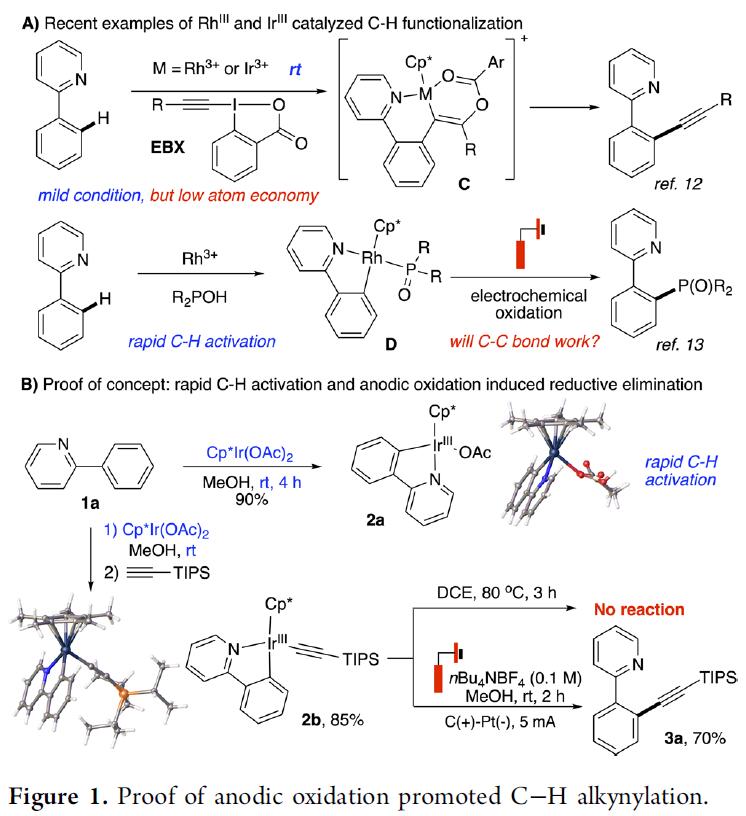
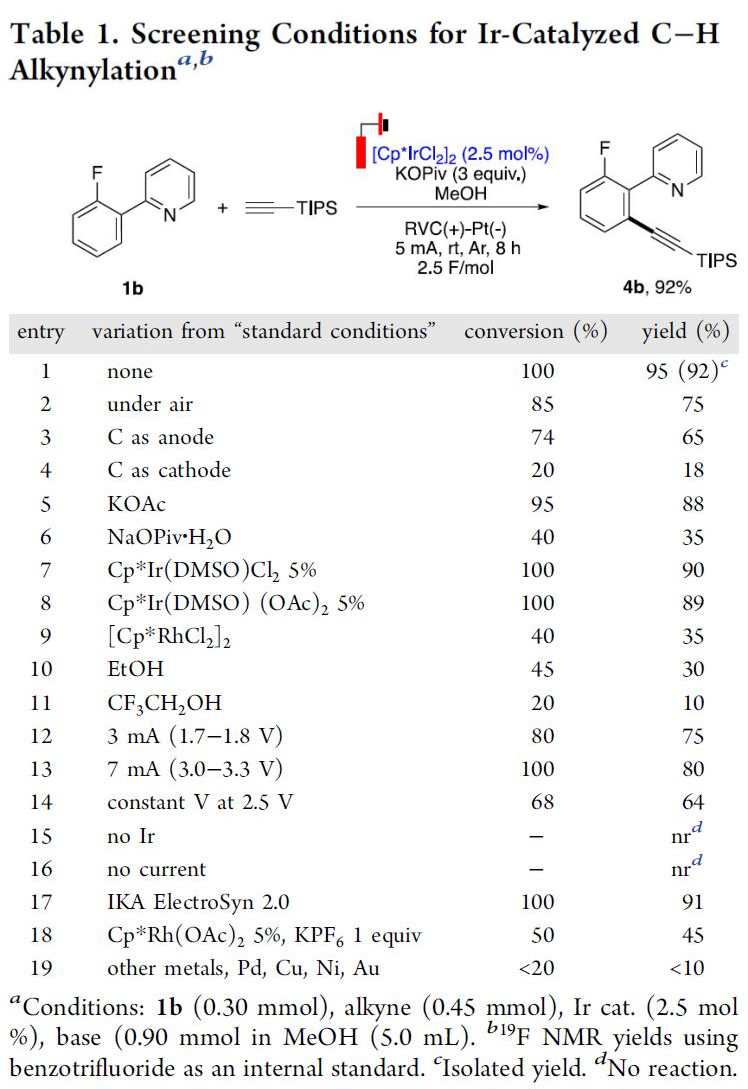
首先,作者对反应条件进行了筛选(Table 1)。反应最佳条件为:使用[Cp*IrCl2]2为催化剂,KOPiv作为碱和电解质,甲醇作为溶剂,可在5.0 mA电流中室温反应(Ar保护),即可获得95%收率的目标产物4b。
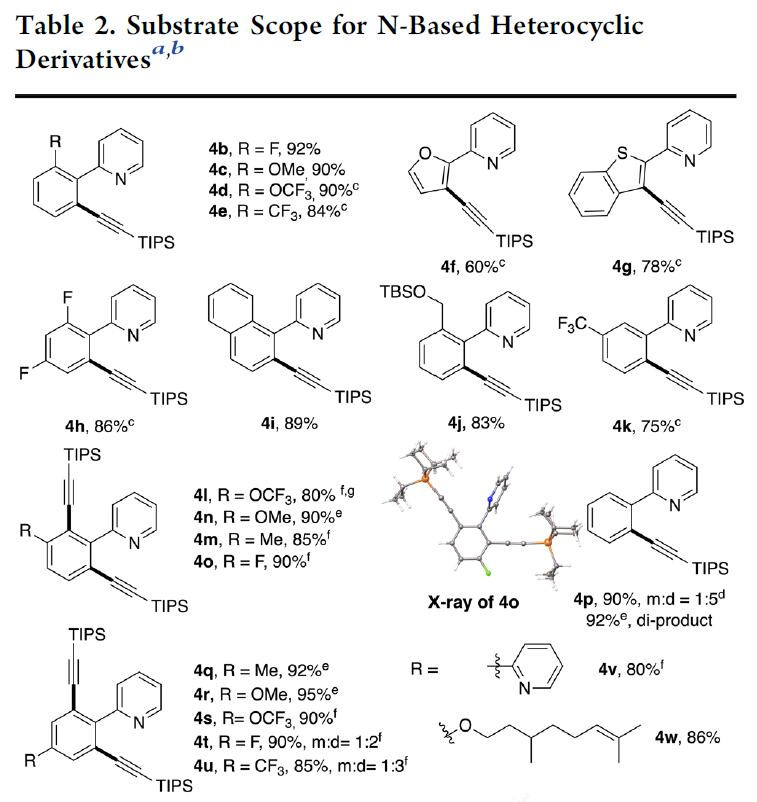
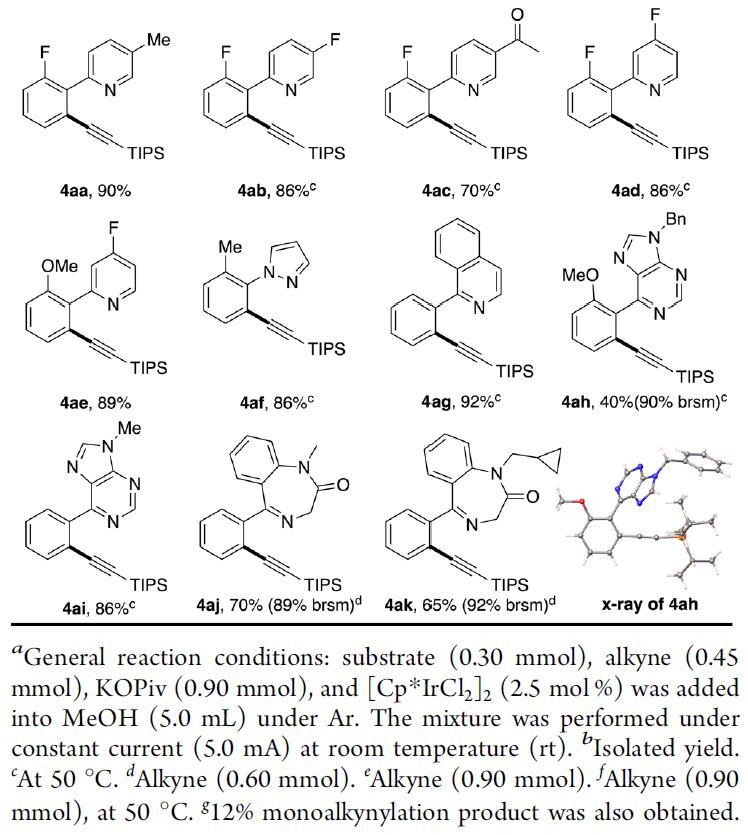
在获得上述最佳反应条件后,作者开始对底物进行扩展(Table 2)。反应结果表明,给电子基团(EDG)和吸电子基团(EWG)取代的芳烃均能很好地发挥作用。通常,EDG取代的底物可在室温下反应,以极高的收率(> 90%)获得所需的产物。在某些情况下,具有EWG取代的底物需要高温(50 oC)才能完全转化(4d,4e)。对于具有较大间位取代基(4k)的苯,具有良好的区域选择性(> 10:1),并且在受阻较小的C-H上发生了炔基化反应。在其他情况下,单-和二-炔基化之间的选择性差(41–4w)。然而,在炔烃过量的情况下,以良好的收率获得二炔基化产物。该策略也适合于空间位阻更大的间位取代苯(41–4o)的二炔基化。同时,具有吡啶(4v)和烯烃(4w)底物也与体系相容。此外,含有EDG和EWG的吡啶环,均以优异的收率获得所需的产物4aa–4ad。吡唑(4af)、喹啉(4ag)和嘌呤(4ah,4ai)也可用作导向基团来完成此转化。值得注意的是,地西泮衍生物(4aj,4ak)也可成功的引入炔基基团,从而说明了该方法在药物分子的后期功能化的潜在应用。
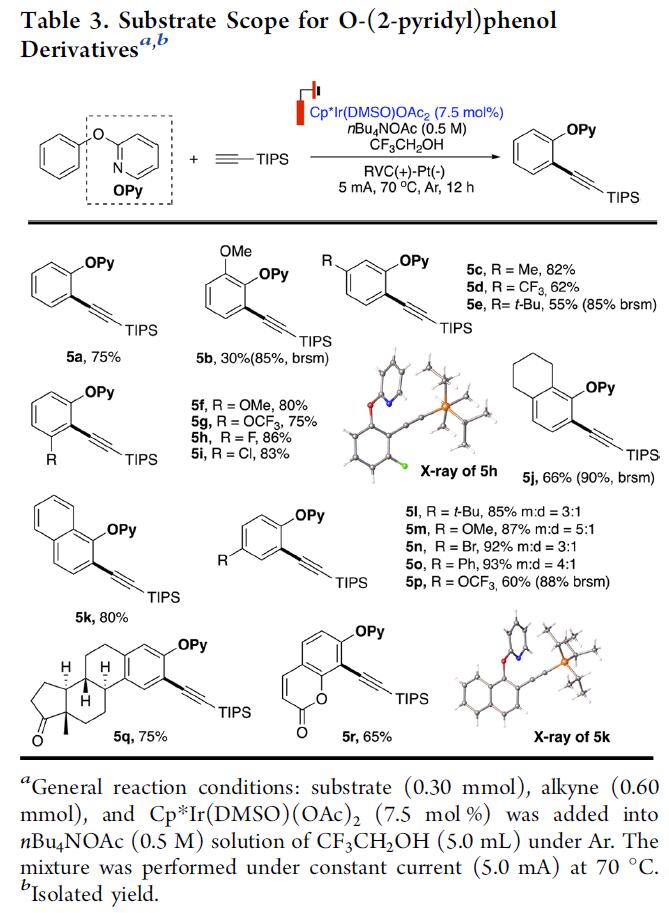
随后,作者对O-(2-吡啶基)苯酚衍生物底物进行了扩展(Table 3)。然而,在上述最佳条件下,O-(2-吡啶基)苯酚衍生物未能实现此类转化。因此,作者重新进行条件的优化,最终确定了使用Cp*Ir(DMSO)(OAc)2作为催化剂(7.5 mol%),nBu4NOAc(0.50 M)作为电解质,在70 ℃的CF3CH2OH中通入5 mA恒定电流,即可获得75%收率的目标产物5a。对于该底物,取代基的位置起关键作用。只带有OPy取代时(5b,5j),转化率略有降低。对于间位含有较大取代基(5c–5e),可以在位阻较小的位置实现炔基化反应。有趣的是,在间为上具有OMe、OCF3、F、Cl等时,则在更受阻碍的C-H键发生炔基化反应,获得反向的区域选择性产物5f–5i,5r。值得注意的是,在对位含有不同的取代基时,只获得单炔基化产物5l–5p,突出了这类特定底物的良好化学选择性。同样,活性的溴基团(5n),含有雌酮(5q)和香豆素(5r)部分的底物,也同样在这种条件下可以耐受。
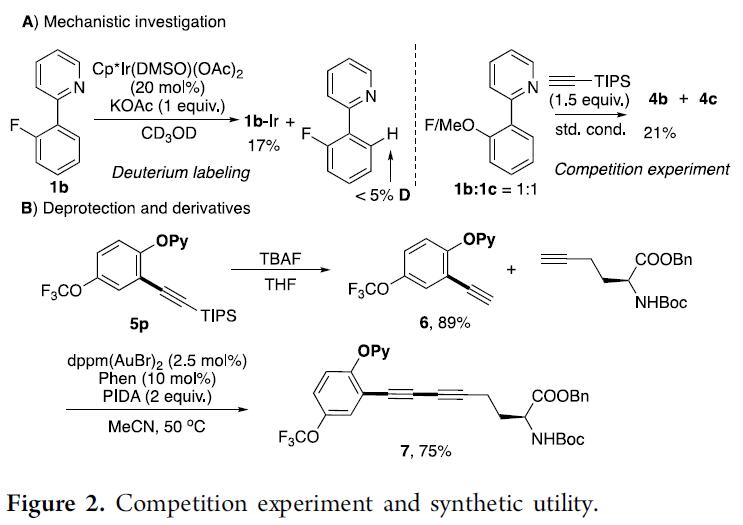
为了进一步了解反应机理,作者首先进行了氘标记实验。与先前报道的Rh催化不同[6],在CD3OD中将1a与Cp*Ir(OAc)2混合时,未观察到H-D交换。该结果表明C-Ir键具有出色的稳定性,这可能导致Ir和Rh金属环之间的反应性不同(Figure 2A)。同时,还进行了含有富电子和缺电子芳环的底物之间的竞争实验。结果表明,富电子的芳环更受青睐,涉及乙酸盐辅助的C-H活化途径(CMD,协同金属化去质子化),然后形成稳定的Ir-C键。此外,根据文献方法[7],吡啶保护基易除去,得到炔基取代的酚。为了进一步证明其合成实用性,作者进行了金催化6与氨基酸修饰的炔烃的氧化偶联,以75%的收率获得氨基酸衍生物7(Figure 2B)。总的来说,该反应在温和条件下具有良好官能团耐受性,同时也为复杂分子的后期官能化提供了有效的途径。
总结
南佛罗里达大学史晓东和衢州学院谢艳课题组共同报道了通过Ir(III)催化经电化学阳极氧化诱导的还原消除,实现与末端炔烃的定向C-H炔基化反应。同时,该反应具有广泛的底物范围、温和反应条件以及出色的官能团耐受性。此外,该反应不仅代表了一种无需外部氧化剂即可引入炔烃官能团的新型原子经济方法,而且还提供了有关Ir(III)促进的C-H活化的机理见解,这将导致新合成用途的发现。
参考文献
- (1) (a) Lyons, T. W.; Sanford, M. S. Palladium-Catalyzed Ligand-Directed C-H Functionalization Reactions. Chem. Rev. 2010, 110, 1147-1169. (b) Colby, D. A.; Tsai, A. S.; Bergman, R. G.; Ellman, J. A. Rhodium Catalyzed Chelation-Assisted C-H Bond Functionalization Reactions. Acc. Chem. Res. 2012, 45, 814-825. (c) Engle, K. M.; Mei, T. S.; Wasa, M.; Yu, J. Q. Weak Coordination as a Powerful Means for Developing Broadly Useful C-H Functionalization Reactions. Acc. Chem. Res. 2012, 45, 788-802. (d) Neufeldt, S. R.; Sanford, M. S. Controlling Site Selectivity in Palladium-Catalyzed CH Bond Functionalization. Acc. Chem. Res. 2012, 45, 936-946. (e) Rouquet, G.; Chatani, N. Catalytic Functionalization of C(sp(2))-H and C(sp(3))-H Bonds by Using Bidentate Directing Groups. Angew. Chem. Int. Ed. 2013, 52, 11726-11743. (f) Chen, Z. K.; Wang, B. J.; Zhang, J. T.; Yu, W. L.; Liu, Z. X.; Zhang, Y. H. Transition metalcatalyzed C-H bond functionalizations by the use of diverse directing groups. Org. Chem. Frontiers 2015, 2, 1107-1295. (g) Daugulis, O.; Roane, J.; Tran, L. D. Bidentate, Monoanionic Auxiliary-Directed Functionalization of Carbon-Hydrogen Bonds. Acc. Chem. Res. 2015, 48, 1053-1064.
- (2) For reviews, see: (a) Wang, M. M.; Wang, Z. X.; Shang, M.; Dai, H. X. Transition-Metal-Catalyzed C-H Alkynylation. Chinese J. Org. Chem. 2015, 35, 570-577. (b) Panda, B. Joy and Challenges of Alkynylation of Arenes and Heteroarenes through Double C-H Functionalizations. Asian J. Org. Chem. 2020, 9, 492-507. (c) Messaoudi, S.; Brion, J. D.; Alami, M. Transition-Metal-Catalyzed Direct C-H Alkenylation, Alkynylation, Benzylation, and Alkylation of (Hetero)arenes. Eur. J. Org. Chem. 2010, 2010, 6495-6516. (d) Dudnik, A. S.; Gevorgyan, V. Formal Inverse Sonogashira Reaction: Direct Alkynylation of Arenes and Heterocycles with Alkynyl Halides. Angew. Chem. Int. Ed. 2010, 49, 2096-2098. (e) Caspers, L. D.; Nachtsheim, B. J. Directing-Group-mediated C−H-Alkynylations. Chem. Asian J. 2018, 13, 1231-1247. For examples of Sonogashira related alkynylation, see: (f) Sonogashira, K.,; Tohda, Y.; Hagihara, N. A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes and Bromopyridines. Tetrahedron Lett. 1975, 16, 4467-4470. (g) Panda, B.; Sarkar, T. K. Gold and palladium combined for the Sonogashira-type cross-coupling of arenediazonium salts. Chem. Commun. 2010, 46, 3131-3133. (h) Chinchilla, R.; Nájera, C. Recent advances in Sonogashira reactions. Chem. Soc. Rev. 2011, 40, 5084-5121.
- (3) (a) Yang, Q.-L.; Xing, Y.-K.; Wang, X.-Y.; Ma, H.-X.; Weng, X.-J.; Yang, X.; Guo, H.-M.; Mei, T.-S. Electrochemistry- Enabled Ir-Catalyzed Vinylic C–H Functionalization. J. Am. Chem. Soc. 2019, 141, 18970-18976. (b) Kong, W.-J.; Finger, L. H.; Messinis, A. M.; Kuniyil, R.; Oliveira, J. C. A.; Ackermann, L. Flow Rhodaelectro-Catalyzed Alkyne Annulations by Versatile C–H Activation: Mechanistic Support for Rhodium(III/IV). J. Am. Chem. Soc. 2019, 141, 17198-17206. For examples of non electrochemical condition, see: (a) Corre, Y.; Werlé, C.; Brelot-Karmazin, L.; Djukic, J.-P.; Agbossou-Niedercorn, F.; Michon, C. Regioselective hydrosilylation of terminal alkynes using pentamethylcyclopentadienyl iridium(III) metallacycle catalysts. J. Mol. Catal. A: Chem. 2016, 423, 256-263. (b) Wang, N. C.; Li, B.; Song, H. B.; Xu, S. S.; Wang, B. Q. Investigation and Comparison of the Mechanistic Steps in the (Cp*MCl2)(2) (Cp*=C5Me5; M = Rh, Ir)-Catalyzed Oxidative Annulation of Isoquinolones with Alkynes. Chem. Eur. J. 2013, 19, 358-364. (c) Peneau, A.; Guillou, C.; Chabaud, L. Recent Advances in Cp*M-III (M = Co, Rh, Ir)-Catalyzed Intramolecular Annulation Through C-H Activation. Eur. J. Org. Chem. 2018, 2018, 5777-5794.
- (4) The example of electrochemical C-H alkynylation/annulation process see: Tian, C.; Dhawa, U.; Scheremetjew, A.; Ackermann, L. Cupraelectro-Catalyzed Alkyne Annulation: Evidence for Distinct C–H Alkynylation and Decarboxylative C–H/C–C Manifolds. ACS Catal. 2019, 9, 7690-7696.
- (5) Xie, F.; Qi, Z. S.; Yu, S. J.; Li, X. W. Rh(III)- and Ir(III)-Catalyzed C-H Alkynylation of Arenes under Chelation Assistance. J. Am. Chem. Soc. 2014, 136, 4780-4787.
- (6) Wu, Z.-J.; Su, F.; Lin, W.; Song, J.; Wen, T.-B.; Zhang, H.- J.; Xu, H.-C. Scalable Rhodium(III)-Catalyzed Aryl C−H Phosphorylation Enabled by Anodic Oxidation Induced Reductive Elimination. Angew. Chem. Int. Ed. 2019, 58, 16770-16774.
- (7) (a) Fan, F.; Tang, J.; Luo, M.; Zeng, X. Chromium-Catalyzed Regioselective Kumada Arylative Cross-Coupling of C(aryl)–O Bonds with a Traceless Activation Strategy. J. Org. Chem. 2018, 83, 13549-13559.(b) Ackermann, L.; Diers, E.; Manvar, A. Ruthenium-Catalyzed C–H Bond Arylations of Arenes Bearing Removable Directing Groups via Six-Membered Ruthenacycles. Org. Lett. 2012, 14, 1154-1157.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!













No comments yet.