
本文作者:杉杉
导读
近日,山东第一医科大学的X. Guan课题组在Green Chem.中发表论文,报道一种无金属条件下的炔基化合物与亚砜之间的氧化还原芳基化反应方法学,进而成功获得一系列γ-芳基化的1,3-二羰基化合物。并且,反应过程中表现出高度的原子经济性。同时,反应机理研究表明,通过原位形成的共轭联烯酮 (conjugated allenone) 中间体,能够较好地解决非对称二烷基取代内炔底物的反应活性以及区域选择性的相关问题,并且能够有效地促进一系列具有吸电子基团取代的炔基底物的官能团化过程。此外,研究发现,通过这一全新的氧化还原芳基化策略形成的各类芳基化1,3-二羰基化合物能够进一步应用于其它不同类型的官能团化过程。
Formal Metal-Free γ-Arylation of 1,3-Dicarbonyl Compounds via an Isomerisation/1,4-Addition/[3,3]-Sigmatropic Rearrangement Sequence
S. Lu, F. Wen, X. Guan, Green Chem. 2021, ASAP. doi: 10.1039/D1GC02856A.
正文
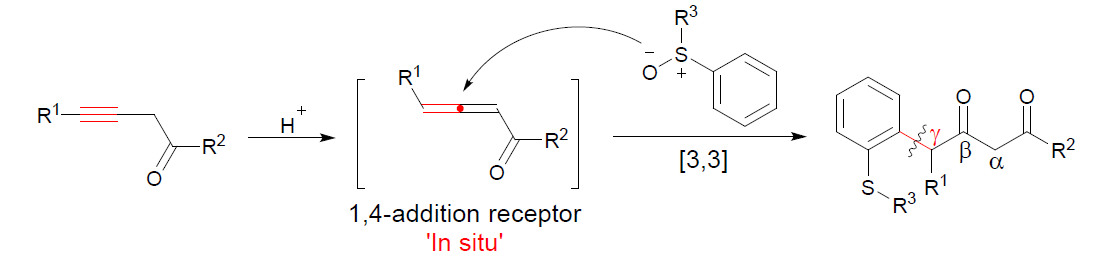
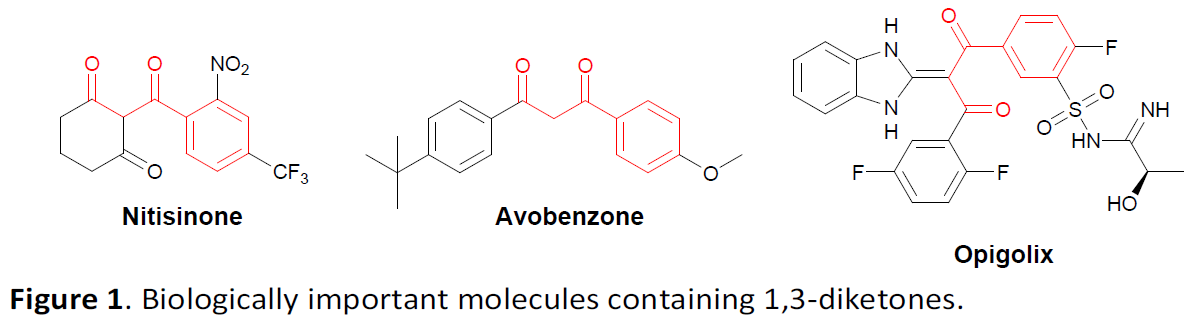
1,3-二羰基化合物作为重要的合成砌块,目前已经成功应用于诸多的有机合成转化过程,例如Knoevenagel缩合、Hantzsch二氢吡啶合成、Tsuji-Trost反应以及DeMayo反应。同时,1,3-二羰基化合物同样能够作为诸多过渡金属催化的有机合成策略研究中的关键配体 (Figure 1)。尽管1,3-二羰基化合物相关的合成方法学研究已经取得诸多进展[1],尤其,1,3-二羰基化合物的直接区域选择性官能团化方法学的相关研究,已经开始受到诸多研究团队的关注。然而,对于1,3-二羰基化合物的γ-官能团化方法学却则较少有相关的研究报道 (Scheme 1a and 1b) [2]–[4]。并且,相关的合成转化研究中通常存在实验操作繁琐、反应条件苛刻、需要采用强碱以及底物应用范围较为有限等弊端。因此,设计一种更为有效并具有高度区域选择性的反应策略,进而成功实现一系列1,3-二羰基化合物的γ-芳基化过程将面临诸多挑战。
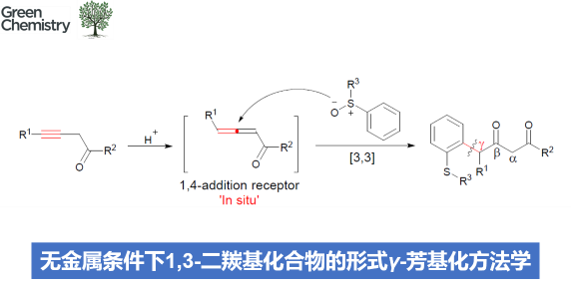
近期,氧化还原中性芳基化反应方法学的相关研究,同样已经取得诸多进展[5]–[8]。然而,对于通过各类β-位置具有吸电子基团取代的非对称炔基化合物参与的芳基化反应方法学,至今尚未有相关的文献报道。尽管根据目前广泛接受的乙烯基正离子机理,涉及上述底物参与的芳基化过程可能较为不利。然而,这里,作者设想,具有吸电子基团取代的炔基化合物,能够通过酸性条件下的异构化过程,原位形成具有良好亲电性的联烯衍生物。之后,进一步通过联烯衍生物与芳基亚砜之间的1,4-加成过程,形成后续[3,3]-σ重排过程的关键中间体 (Scheme 1c),进而显著提升二烷基取代炔基化合物参与相关反应过程时的区域选择性。同时,能够进一步提高非活性炔基化合物的反应活性,进而顺利完成各类非对称二烷基取代内炔化合物参与的氧化还原中性芳基化过程,并获得一系列相应γ-芳基化的1,3-二羰基产物。基于上述的研究设想,本文,作者成功设计出一种在无金属试剂存在的条件下,采用1,3-二羰基化合物参与的形式γ-芳基化方法学。
首先,作者采用1a与2a作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用TfOH作为催化剂,乙酸乙酯作为反应溶剂,反应温度为室温。最终以高度的区域选择性以及92%反应收率,获得相应的γ-芳基化产物3a。
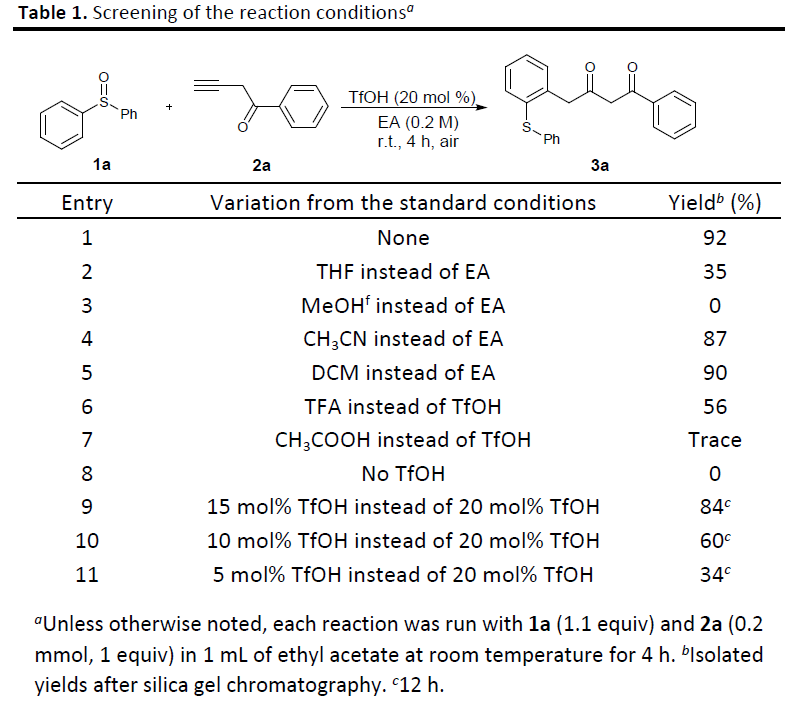
在上述最佳反应条件下,作者对各类炔酮底物的应用范围进行考察 (Scheme 2)。研究表明,在芳环不同位置中具有一系列供电子与吸电子基团取代的芳基炔酮底物,均能较好地与上述反应条件兼容,并获得相应的γ-芳基化产物3b–3s (71-94% 反应收率)。之后,作者发现,炔酮底物中的R2取代基团为萘基、苯乙烯基、杂芳基以及烷基时,同样能够顺利完成上述的γ-芳基化过程,并获得相应的目标产物3t–3z (70-95% 反应收率)。同时,该小组发现,对于炔基α-位具有酯基取代的炔酮底物,则需要更高的反应温度,并且,同样能够获得相应的γ-芳基化产物3a’与3b’。然而,研究表明,上述的标准反应条件对于具有酰胺基团取代的炔酮底物,则无法获得预期的γ-芳基化产物3c’。
接下来,作者进一步对一系列亚砜底物的应用范围进行考察。研究表明,一系列不同类型的亚砜底物,均能够顺利地参与上述的γ-芳基化过程,并获得相应的目标产物3d’–3h’ (84-91% 反应收率)。同时,研究发现,亚砜与内炔基化合物之间的反应,则需要较长的反应时间,最终同样能够以优良的反应收率与区域选择性,获得相应的支链产物3i’与3j’。接下来,该小组进一步发现,这一全新的γ-芳基化策略同样能够有效地应用于一系列高度官能团化的药物分子的合成,例如Fexinidazole与Sulindac (Scheme 2, 3k’与3l‘)。
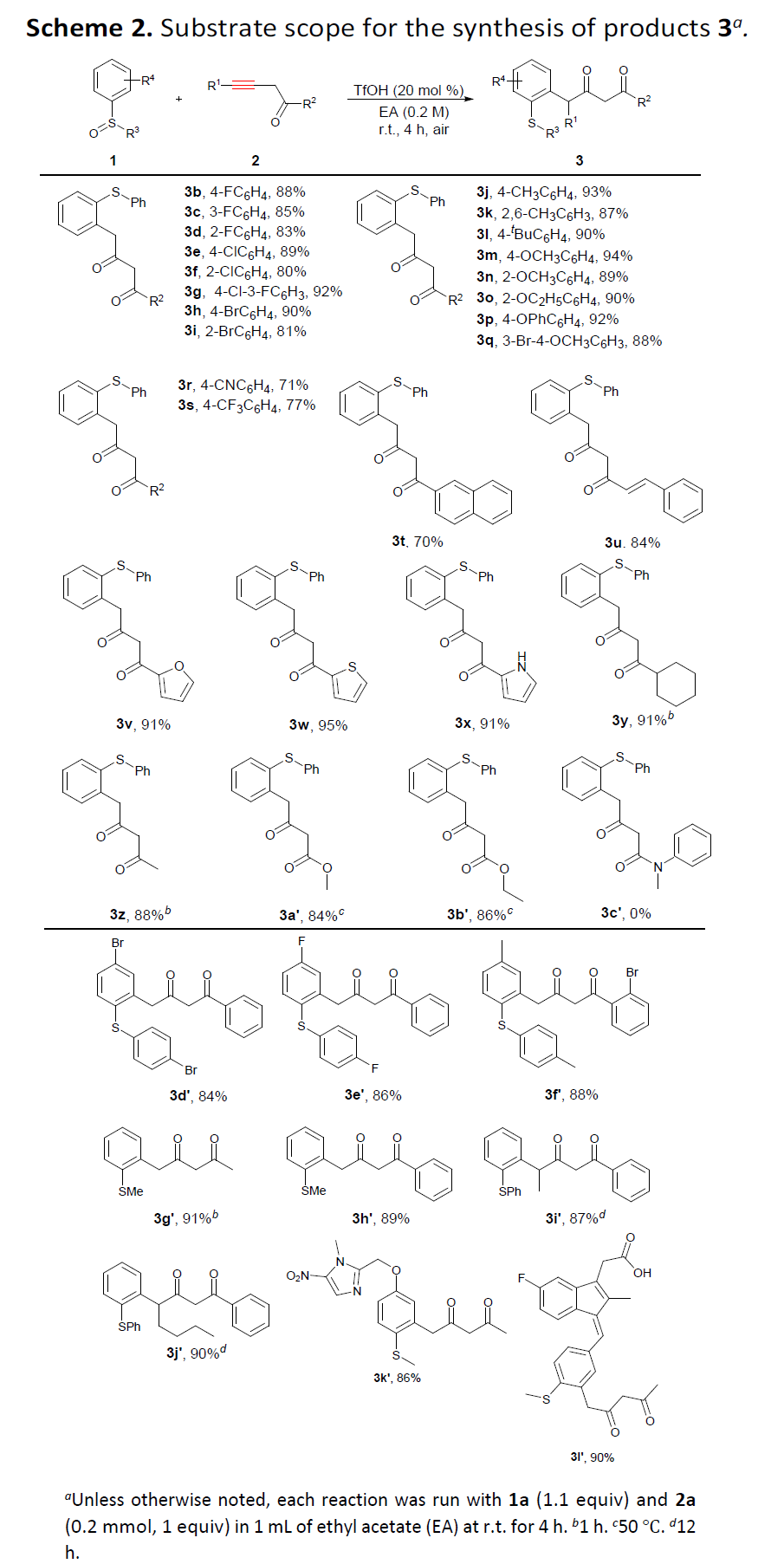
之后,为提出合理的反应机理,作者进行一系列相关的控制实验研究 (Scheme 3)。首先,该小组发现,采用共轭联烯酮4在上述的标准条件下与1a反应时,能够获得94%收率的γ-芳基化产物3a。进而表明共轭联烯酮作为上述γ-芳基化过程的关键中间体 (Scheme 3a)。之后,该小组发现,在无TfOH存在时,共轭联烯酮4的γ-芳基化过程无法顺利进行 (Scheme 3b)。同时,作者假设,酸的存在不仅能够促进相应炔酮底物的异构化,形成共轭联烯酮4的反应过程。并且,同样能够促进上述γ-芳基化过程的有效进行。接下来,该小组进一步观察到,在通过炔酮5进行的氘标记实验中,能够获得羰基γ-位置具有氘取代的芳基化产物6 (Scheme 3c)。基于上述的实验研究,作者提出一种合理的机理路径 (Scheme 3d)。首先,通过酸性条件下,α-羰基炔底物2的异构化过程,形成具有较高反应活性的共轭联烯酮中间体III。之后,通过芳基亚砜对于共轭联烯酮III中β-位置的Michael加成过程,产生中间体IV。并通过中间体IV经历后续的[3,3]-σ重排过程,获得预期的γ-芳基化产物3。
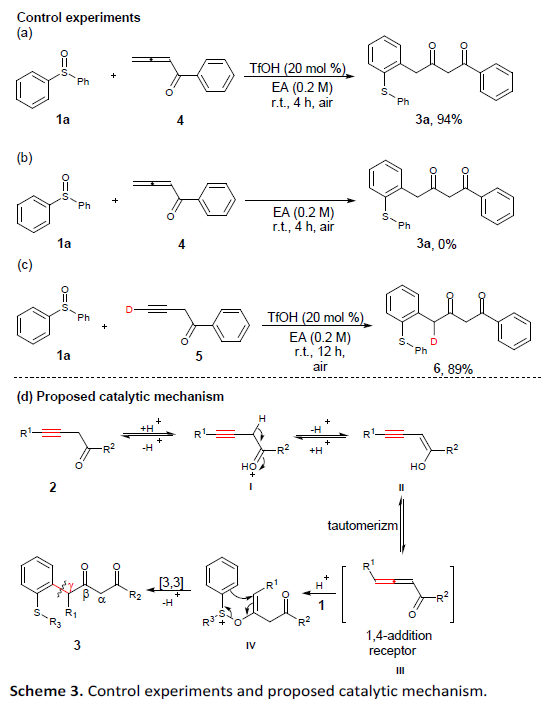
同时,该小组进一步对上述γ-芳基化方法学的合成应用价值进行深入研究(Scheme 4)。首先,作者发现,将底物1e’的用量扩大至20 mmol时,同样能够成功获得86%收率的目标产物3e’。并且,该小组进一步发现,通过3e’作为关键砌块,能够进行多种不同类型的合成转化过程,进而成功实现一系列具有重要应用价值的杂环分子的构建。
总结
本文中,作者成功设计出一种构建γ-芳基化的1,3-二羰基化合物的有效策略。同时,通过反应机理的相关研究表明,通过原位形成的共轭联烯酮中间体,能够有效地解决各类烷基取代炔基化合物的反应活性以及区域选择性的相关问题。同时,能够顺利完成一系列具有吸电子基团取代的炔基衍生物的官能团化过程。并且,这一全新的γ-芳基化策略具有实验操作简便、反应条件温和、广泛的底物应用范围、高度的原子经济性以及优良的区域选择性控制等优势。
参考文献
[1] (a) Z. He, X. Qi, Z. She, Y. Zhao, S. Li, J. Tang, G. Gao, Y. Lan, J. You, J. Org. Chem. 2017, 82, 1403. doi: 10.1021/acs.joc.6b02575.(b) J. Kuang, T. Zhou, T. You, J. Chen, C. Su, Y. Xia, Org. Biomol. Chem. 2019, 17, 3940. doi: 10.1039/C9OB00494G.
(c) G. You, Z. Chang, J. Yan, C. Xia, F. Li , H. Li, Org. Chem. Front. 2021, 8, 39. doi: 10.1039/D0QO01174F.
(d) J. Chen, D. Joseph, Y. Xia, S. Lee, J. Org. Chem. 2021, 86, 5943. doi: 10.1021/acs.joc.0c02868.
[2] (a) C. R. Hauser, T. M. Harris, J. Am. Chem. Soc. 1958, 80, 6360. doi: 10.1021/ja01556a049.(b) K. G. Hampton, C. R. Hauser, J. Org. Chem. 1965, 30, 2934. doi: 10.1021/jo01020a013.
(c) J. F. Wolfe, T. M. Harris, C. R. Hauser, J. Org. Chem. 1964, 29, 3249. doi: 10.1021/jo01034a029.
[3] K. G. Hampton, T. M. Harris, C. R. Hauser, J. Org. Chem. 1964, 29, 3511. doi: 10.1021/jo01035a015. [4] F. Wagner, K. Harms, U. Koert, Org. Lett. 2015, 17, 5670. doi: 10.1021/acs.orglett.5b02952. [5] D. Kaiser, I. Klose, R. Oost, J. Neuhaus, N. Maulide, Chem. Rev. 2019, 119, 8701. doi: 10.1021/acs.chemrev.9b00111. [6] B. Lu, Y. Li, Y. Wang, D. H. Aue, Y. Luo, L. Zhang, J. Am. Chem. Soc. 2013, 135, 8512. doi: 10.1021/ja401343p. [7] D. Kaiser, L. F. Veiros, N. Maulide, Chem. – Eur. J. 2016, 22, 4727. doi: 10.1002/chem.201600432. [8] A. Pons, J. Michalland, W. Zawodny, Y. Chen, V. Tona, N. Maulide, Angew. Chem., Int. Ed. 2019, 58, 1730.doi:10.1002/anie.201909381.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载













No comments yet.