
本文作者:杉杉
导读
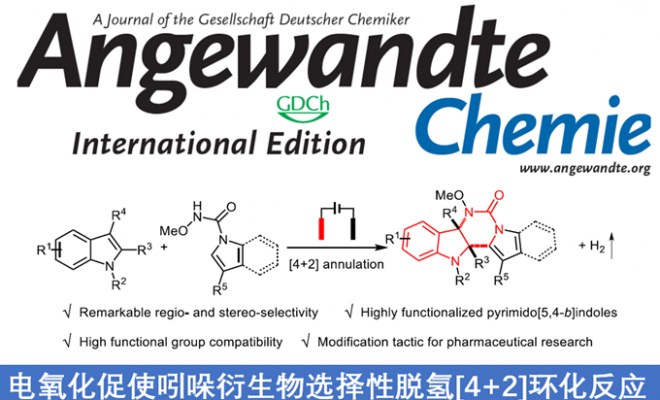
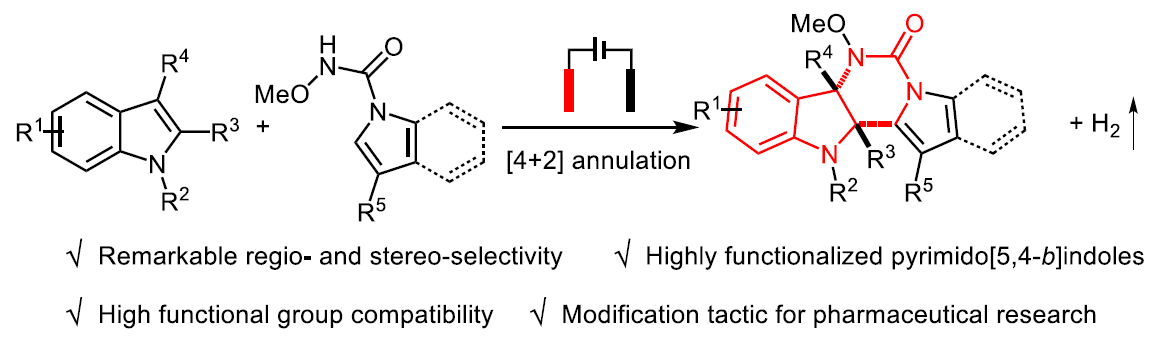
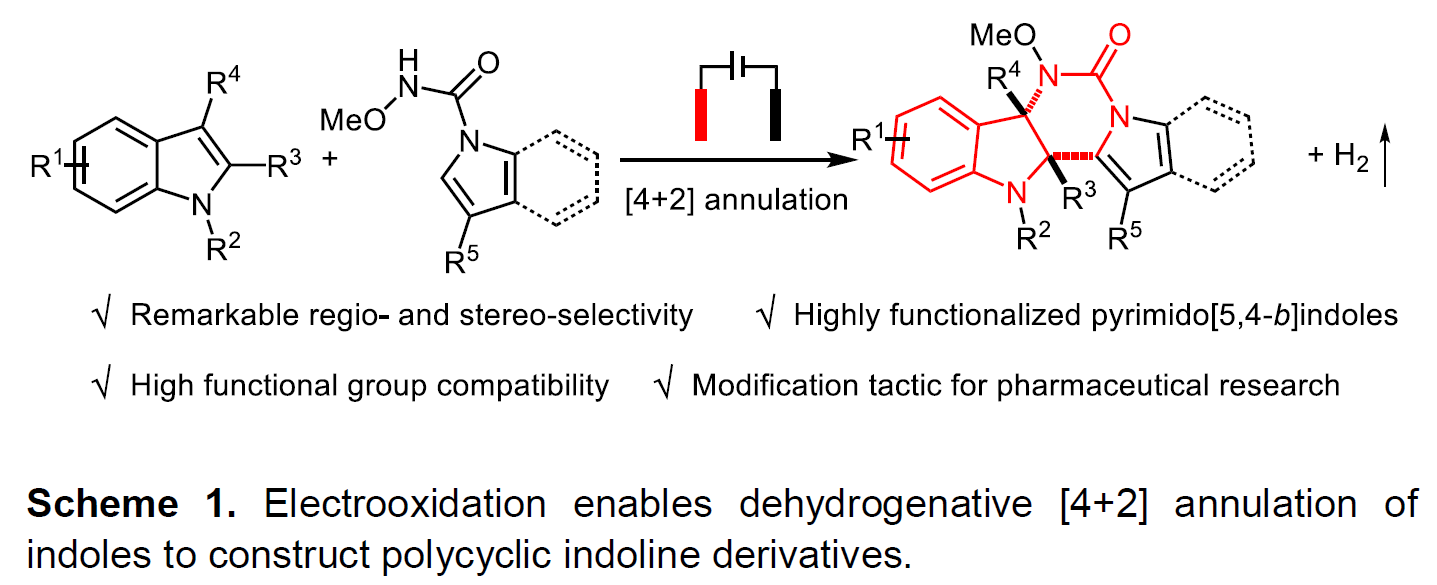
吲哚的去芳化环化反应已成为制备多环吲哚啉生物碱的高效工具。在过去,五元环稠合二氢吲哚的合成方法已被大量报道,但对于六元环稠合二氢吲哚的合成研究却很少。近日,武汉大学雷爱文教授课题组在德国应化杂志发表论文,提出了通过电化学途径,可实现不同吲哚之间的去芳化[4+2]环化反应,具有出色的区域和立体选择性,同时可在无氧化剂和无金属的条件下,获得高度官能化的嘧啶并[5,4-b]吲哚衍生物。值得注意的是,这种电化学方法能够保持优异的官能团耐受性,并可对药物进行相关的后期修饰。初步机理研究表明,在原位产生的吲哚自由基阳离子和以N为中心的自由基之间,通过自由基自由基交叉偶联进行电氧化环化反应。
Electrooxidation Enables Selective Dehydrogenative [4+2] Annulation between Indole Derivatives
Chunlan Song, Kun Liu, Xu Jiang, Xin Dong, Yue Weng Chien-Wei Chiang, and Aiwen Lei
Angew. Chem. Int. Ed. ASAP DOI:10.1002/anie.202000226
正文
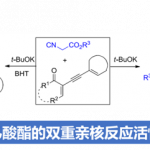
在过去的几十年中,吲哚的去芳化环化反应作为合成多环吲哚啉的生物碱(具有多种生物活性)有效途径。根据C2-C3的π键或吲哚固有的亲核性,可通过1,3-偶极环加成反应、环丙烷化反应、串联亲电加成/环化反应等,均可合成五元环稠合2,3-二氢吲哚衍生物。然而,一些更为重要的六元环稠合2,3-二氢吲哚却很少被研究。在众多可行的方法中,[4+2] Diels-Alder反应作为最常用的方法,其中吲哚可以同时充当两个或四个碳原子的合成子。虽然该方法具有高效性,但由于Diels-Alder反应的固有性质(在N1和C3位置需具有特定吸电子基团或在C2或C3位置具有共轭双键),因此从市售的吲哚进行[4+2]去芳化环化反应的尝试很少成功。此外,也可通过吲哚化合物的氧化诱导实现C-H官能化反应。如Ma课题组通过吲哚分子内的氧化偶联,对多环吲哚啉生物碱进行了全合成研究。You课题组通过使用氧化剂,在可见光诱导下实现吲哚化合物的分子内去芳化反应。尽管上述报道已取得一定的成果,但吲哚化合物的分子间[4+2]环化却很少研究。同时,该类反应均使用化学氧化剂,导致较低的原子经济性以及存在官能团耐受性问题。因此,避免氧化剂的使用,并且从易得的吲哚化合物经分子间[4+2]环化合成六元环稠二氢吲哚衍生物的方法有待开发。
基于课题组前期的研究成果以及电化学合成策略的应用(无需使用氧化剂),作者发现吲哚化合物可以在阳极氧化条件下,在原位生成自由基阳离子中间体。由于吲哚自由基阳离子具有独特的反应性,作者试图引入一种试剂(具有单独亲核中心的自由基)与自由基阳离子进行自由基偶联反应,即可获得所需的[4+2]环化产物。经过大量底物的筛选,作者选择了吲哚-1H-羧酰胺(通常在过渡金属催化的交叉偶联反应中用作导向基团)作为最佳偶联底物。在电解条件下,作者通过与吲哚-1H-羧酰胺混合发现了BHT的胺化产物,表明吲哚-1H-羧酰胺可以作为氮自由基的前体。结合吲哚环固有的强亲核性,作者推测吲哚-1H羧酰胺可同时满足含有自由基和亲核中心的要求。
基于上述的总结,作者提出了一种通过电化学氧化方法,实现吲哚-1H-羧酰胺和1-烷基吲哚的分子间脱氢[4+2]环化反应,合成六元环稠合二氢吲哚衍生物(Scheme 1)。该反应实现了氮自由基参与的分子间脱氢环化反应,而大多数电化学此类文献主要研究分子内的反应。同时,获得多种高度功能化和出色区域选择性的嘧啶并[5,4-b]吲哚衍生物(具有多种生物学和药学活性,如抗HIV-1抑制剂、神经激肽受体和α1-肾上腺素受体等)。
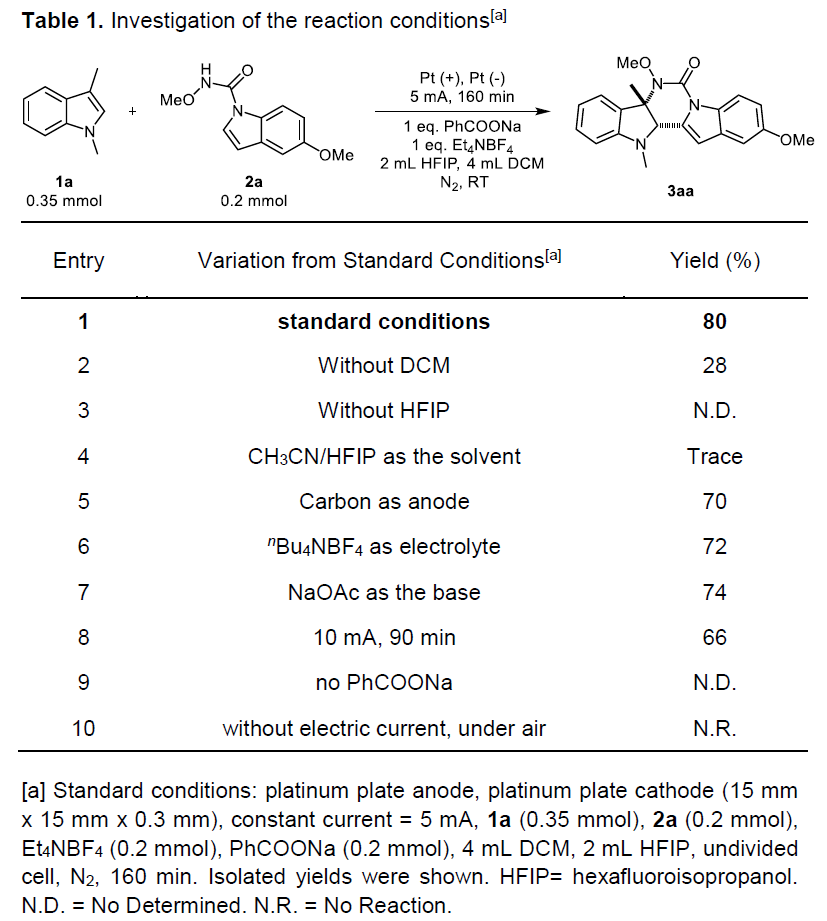
首先,作者以1,3-二甲基吲哚1a和N,5-二甲氧基吲哚-1H-羧酰胺2a作为模型底物,进行了相关 [4+2]环化反应条件的筛选(Table 1)。当在一个简单的无隔膜电解槽(5mA恒定电流)的标准条件下反应,可以获得80%产率的嘧啶并[5,4-b]吲哚产物3aa(entry 1)。此外,当HFIP用作单一溶剂时,[4+2]环化效率较差,而不使用HFIP时,则无法获得目标产物(entries 2-3)。而用CH3CN代替DCM不利于环化(entry 4)。同时,作者对电化学的相关参数也进行了筛选,如电极材料和电解质,当使用碳棒作为阳极会在一定程度上降低产率(entry 5),当使用nBu4NPF6作为电解质可获得良好的产率(entry 6)。通过使用NaOAc代替PhCOONa,以74%的产率获得嘧啶并[5,4-b]吲哚产物3aa(entry 7)。如果将电流调节至10 mA会导致产率略低(entry 8)。最后,对照实验表明,在没有碱或电流的情况下,没有获得任何目标产物(entries 9-10)。
在获得上述最佳反应条件后,作者开始对吲哚底物1进行了扩展(Table 2)。反应结果表明,吲哚的苯环不受电子效应和定位效应的影响,均可获得相应的产物(3bb–3ea),甚至强吸电子基团(CF3)也可获得产物3fc。另外,C-3位具有醚或甲硅烷基醚取代基时,也可进行环化反应获得产物3ga和3ha,同时在温和的电解条件下,游离羟基不受反应的影响,获得含有羟基的产物3ia。具有空间位阻的2,3-二取代的吲哚,也可获相应的产物3ja。此外,吲哚的氮原子上的甲基可以被苄基、烯丙基、氰基和叠氮基取代,分别获得相应的产物3ka–3na。
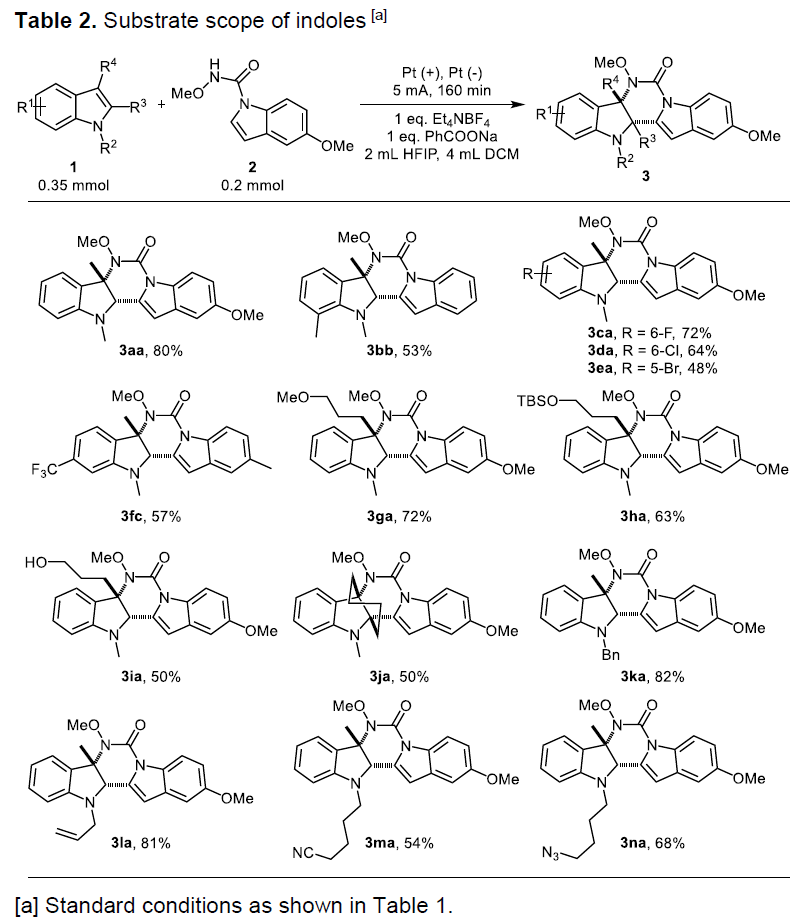
随后,作者对吲哚-1H-羧酰胺底物2进行了相关的扩展(Table 3)。不同的官能团均以中等至良好的产率获得相应的产物3ab–3ag,如果苯环含有强吸电子基团(CF3)时,可获得88%产率的3ah。如果使用含有炔基取代底物时,可获得含炔基的产物3ai,并为炔基的进一步修饰提供了多种可能。同时,也可在产物中引入一些杂芳香基基团,如噻吩(3aj)和二苯并呋喃(3ak)。此外,N-乙氧基酰胺取代的吲哚也可高效的获得环化产物3al。最后,作者也尝试了N-甲氧基-1H-吡咯-1-羧酰胺作为氮源的可能性,分别获得四环稠合嘧啶并[5,4-b]吲哚产物3am(收率77%)和3an(收率53%)。
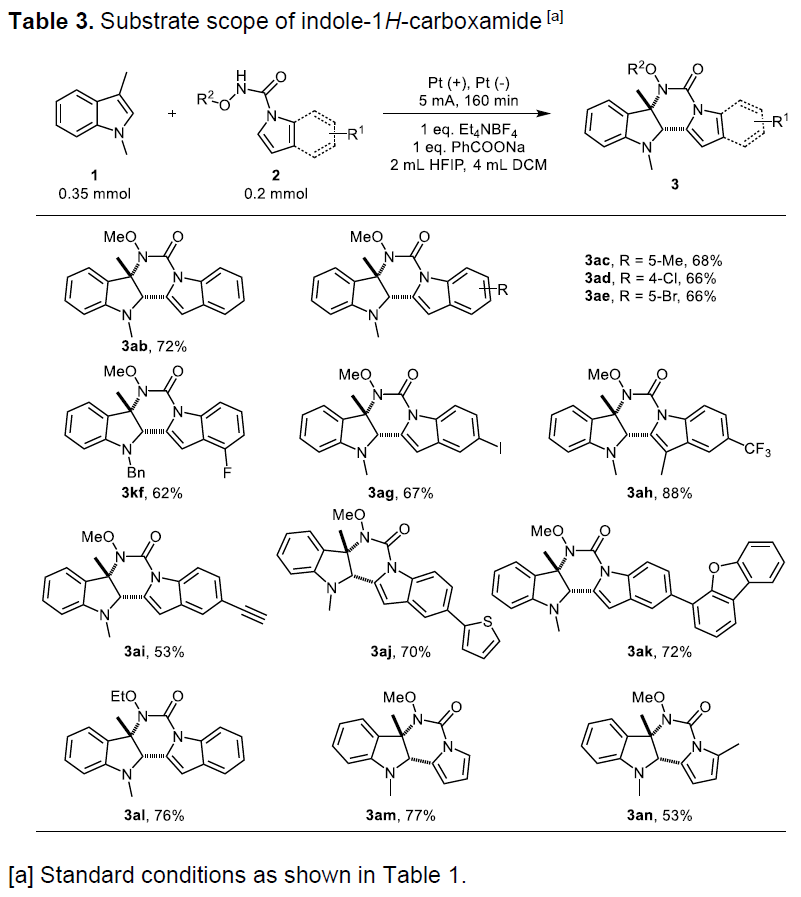
紧接着,作者对底物进行了相关的修饰后可直接合成更具价值的药物和天然产物,从而进一步证明该反应实用性(Table 4)。该反应可使含蛋氨酸的反应物进行官能化,从而表现出了该方法在生物反应中的潜力(5aa)。而在简单的修饰N-甲氧基酰胺基团步骤之后,具有抗糖尿病活性的褪黑激素(melatonin)可以89%的产率参与环化反应(5ab)。此外,当使用洛索洛芬(loxoprofen,一种止痛,解热和消炎剂)、丙磺舒(probenecid,原型尿酸药物)和含扎托洛芬的(zaltoprofen,非甾体类抗炎药)的N-甲氧基酰胺反应物与1,3-二甲基吲哚在标准条件下反应时,分别获得所需的环化产物5ac(84%),5ad(75%)和5ae(69%)。
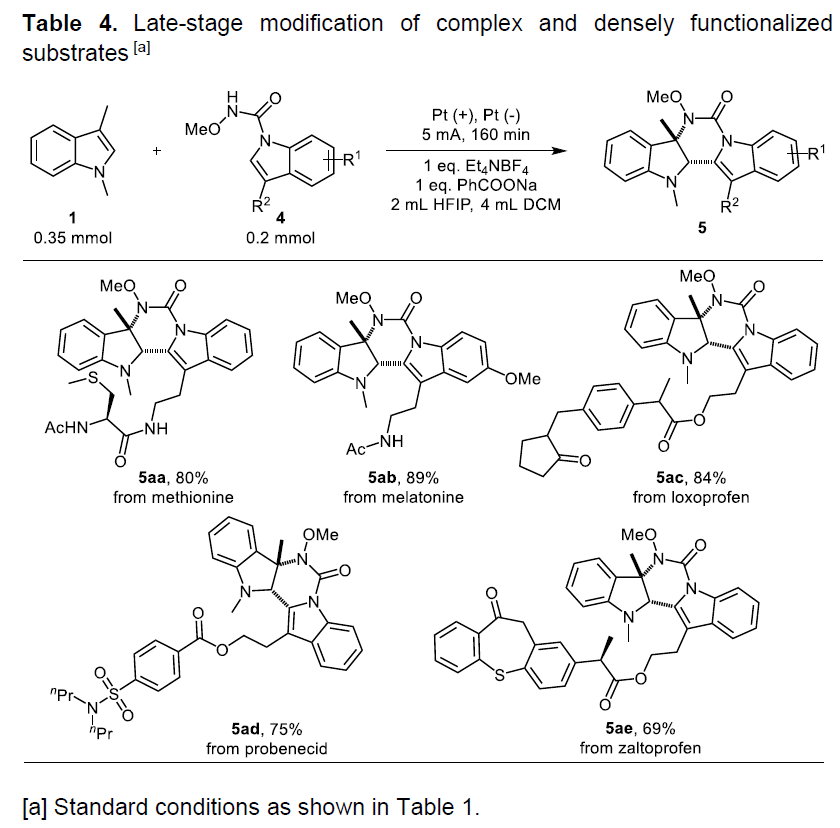
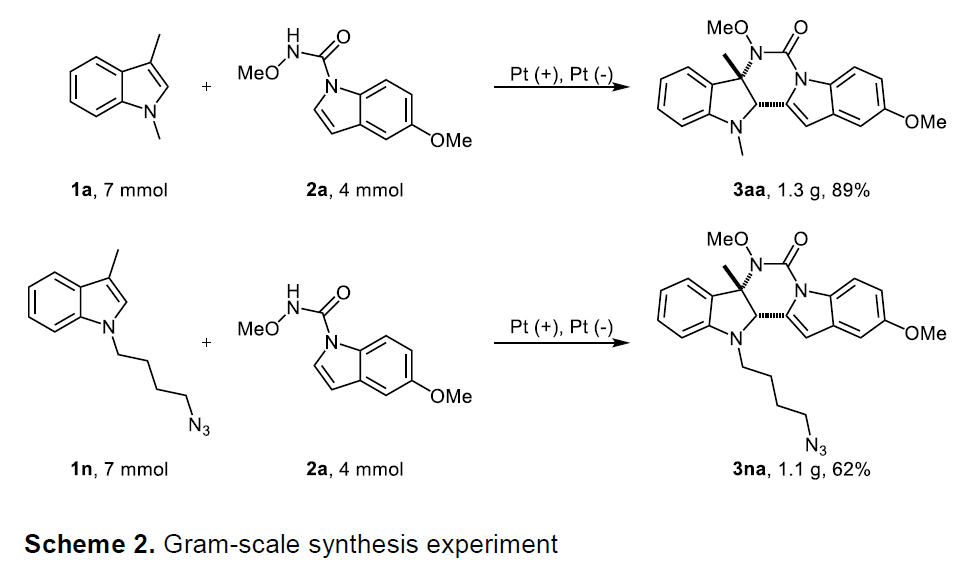
随后,作者也进行了相关的克级反应(Scheme 2),反应结果(选择性和产率)与小试结果基本一致,从而为进一步放大生产提供了可能。
此外,也可以通过简单的操作将高度官能化的嘧啶并[5,4-b]吲哚转化为更具价值的合成中间体或药物(Scheme 3)。首先,N-OMe保护的3ab在室温条件下,使用SmI2进行脱保护,获得95%收率的6a。此外,可使用含有叠氮化物(3na)或碘基团(3ag)二氢吲哚衍生物,与药物分子中的炔基进行Sonogashira偶联反应,分别获得产物6b(90%),6c(93%)和6d(91%)。
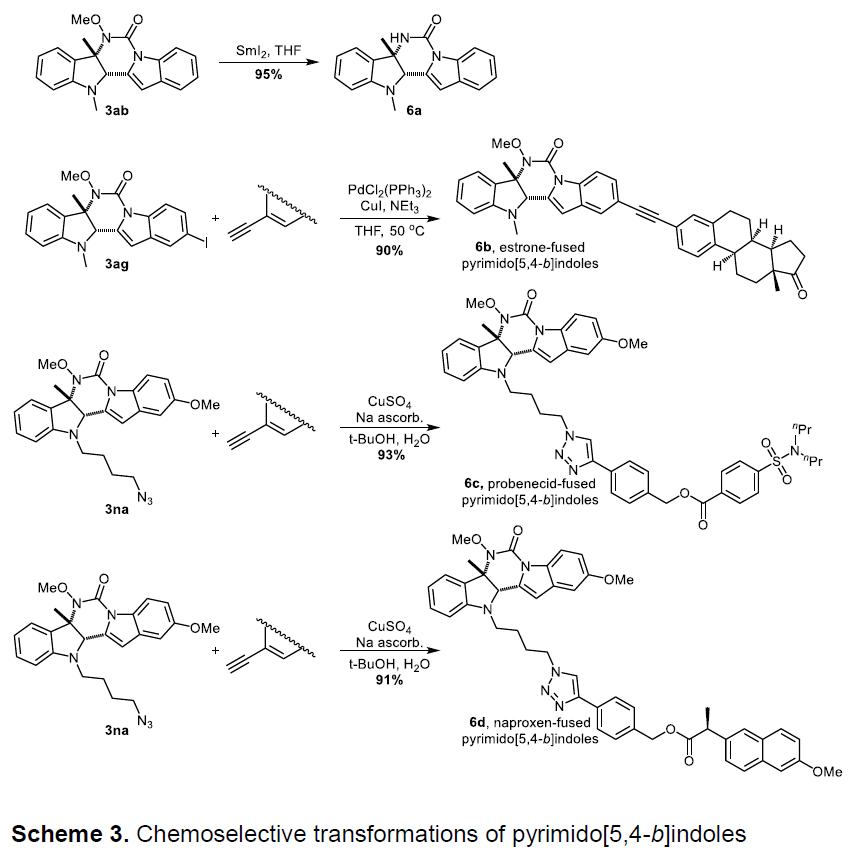
为了进一步了解反应的机理,作者通过对两种标准底物进行循环伏安(CV)实验(Scheme 4)。1,3-二甲基吲哚(1a)约在0.6 V时开始被氧化,而N,5-二甲氧基吲哚-1H-羧酰胺(2a)约在1.1 V开始被氧化。此外,在2a中加入一个当量的PhCOONa,可观察到一个新的氧化峰,约在0.6V开始氧化。这些结果表明,使用化学计量碱可使1a和2a同时被氧化生成具有反应活性的中间体。
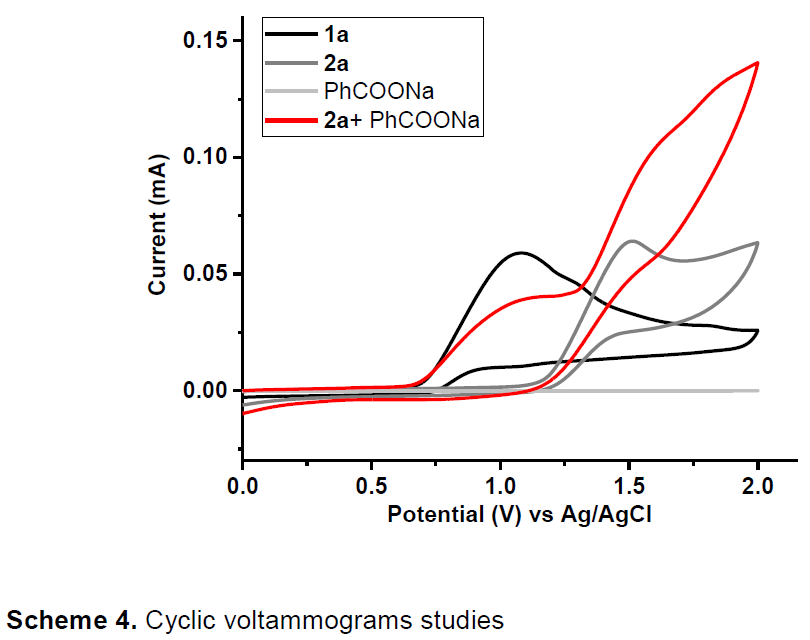
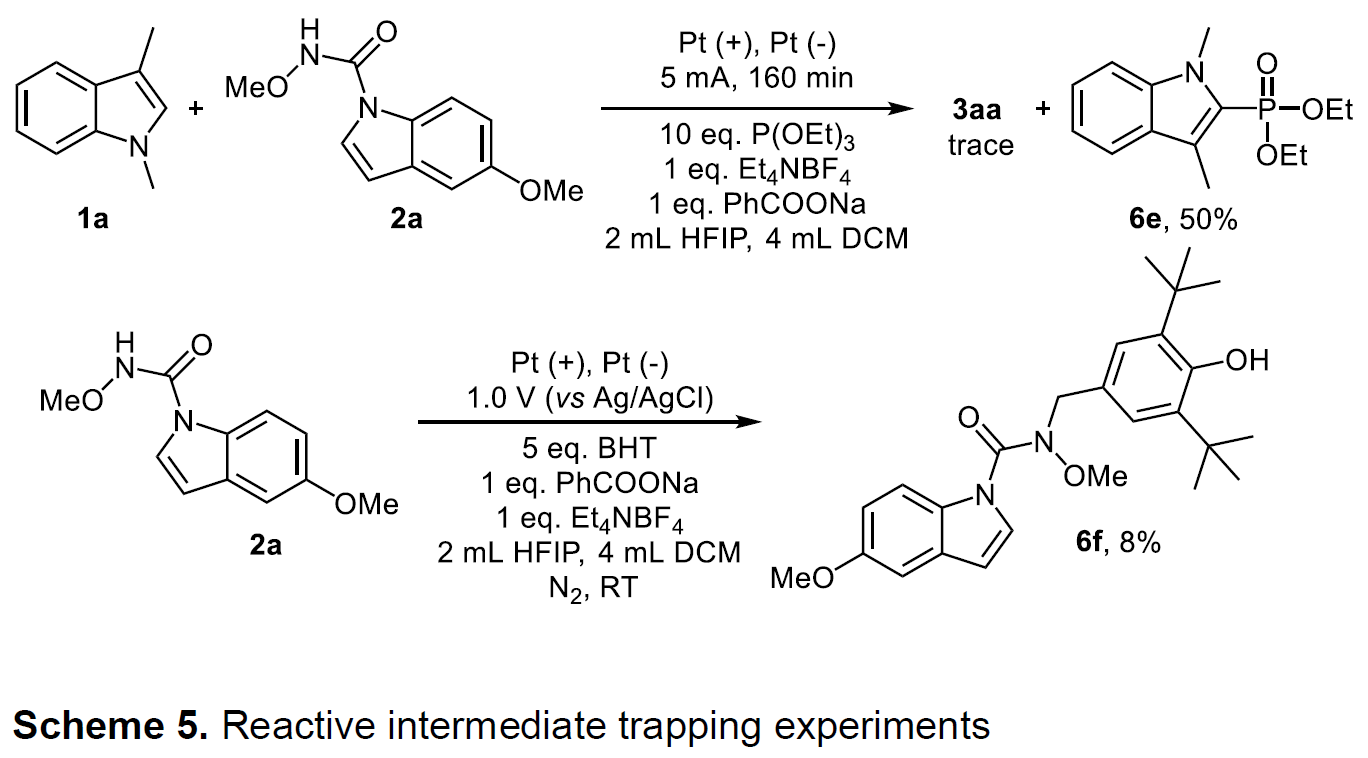
同时,为了确定1a和2a的氧化种类,作者进行了电子顺磁共振(EPR)实验。在存在PhCOONa时,未观察到1a的信号峰,而观察到2a的自由基信号峰。而使用P(OEt)3作为自由基清除剂进行了自由基捕获实验时(Scheme 5),仅获得痕量的环化产物,可分离出吲哚磷酸化产物6e,产率为50%,这表明电解过程中存在1a的吲哚自由基阳离子。另外,将2a与BHT处于恒定电压条件下,获得苄基胺化产物6f。因此,在PhCOONa的作用下,2a也可被氧化生成具有N中心自由基。
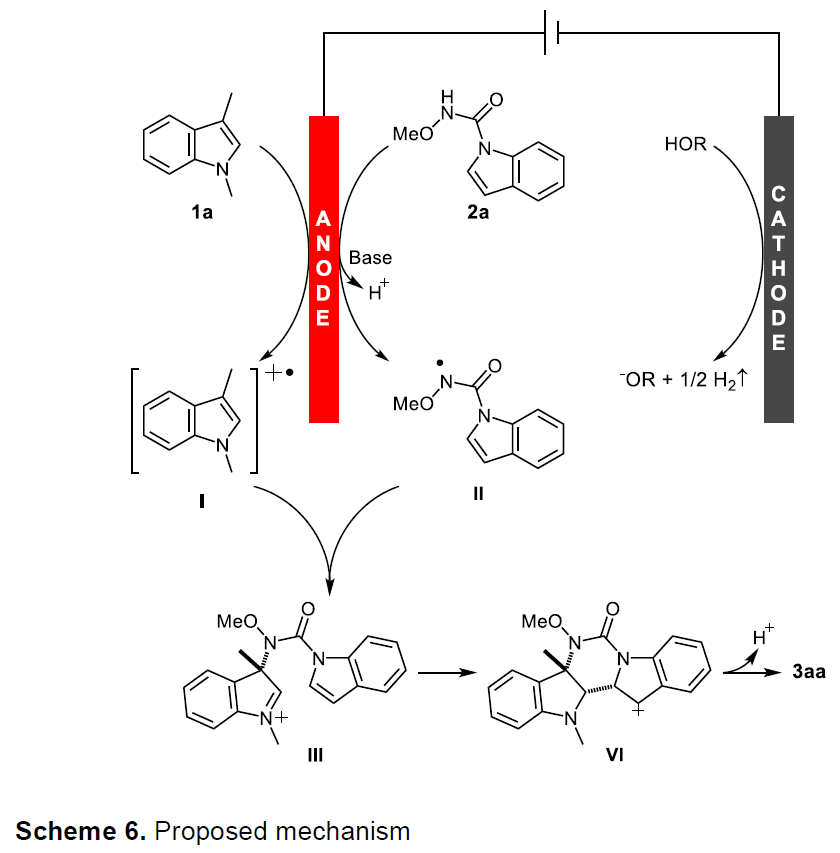
根据上述的实验和相关文献的查阅,作者提出了一种可能的反应机理(Scheme 6)。首先,1a在阳极被氧化生成吲哚自由基阳离子I,而2a在碱的促进下通过单电子转移(SET)氧化获得具有N中心自由基II。随后,吲哚自由基阳离子I和N中心自由基II在C3位之间的实现自由基自由基交叉偶联,获得中间体III。最后,经分子内环化和去质子化后,获得目标产物3aa,而HFIP的阴极经还原反应释放出氢气。
总结
武汉大学雷爱文教授课题组报道了通过电化学途径,实现不同吲哚之间的去芳化[4+2]环化反应,获得高度官能化的嘧啶并[5,4-b]吲哚衍生物。同时避免了氧化剂和金属催化剂的使用,具有出色的区域和立体选择性、官能团耐受性等优点,同时通过对相关底物的修饰可获得多种药物及其天然产物。初步机理研究表明,在原位产生的吲哚自由基阳离子和以N为中心的自由基之间,通过自由基自由基交叉偶联实现环化过程。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!


















No comments yet.