
投稿作者alberto-caeiro
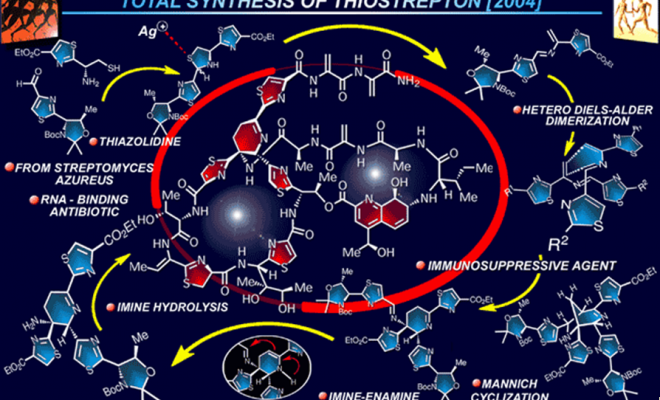
Thiostrepton全合成
1)脱氢呱啶碎片38的合成
脱氢呱啶碎片38的合成是thiostrepton全合成的关键,同时也是全合成中最难的部分,全合成工作自然从它的合成开始。首先是二聚前体42的合成。L-苏氨酸衍生物43经混酸酐得到对应的酰胺,在Lawesson试剂作用下[1],得到硫酰胺44;亲核试剂44与溴化物45反应经中间体46得到羟基噻唑啉47,随后脱水得到噻唑48,此合成噻唑的方法称为Hantzschthiazole synthesis[2]。DIBAL-H将还原酯基后得到醛49。
L-半胱氨酸衍生物50经和HOSu反应得的到相应羧酸酯得到酰胺,随后同样的与Lawesson试剂反应得到硫酰胺51,之后经Hantzsch硫唑合成法得硫唑52;TFA条件下脱除对酸敏感的保护基得到氨基硫醇53;氨基硫醇53与醛49在弱碱性条件下缩合得到二聚前体42。

图8:Synthesis of monomeric thiazolidine derivative 42
在关键的脱氢呱啶的合成中,首先用到的是条件A,虽然得到了期望的产物39,但是是1:1的异构体,并且产率只有22%,主要的产物是桥环化合物57及其对映体57’,这在Wulff的工作中有所体现。通过对主要产物的产生机理分析,发现此串联反应的前面步骤是十分快速的,并且有很简单高效的方法得到期望的产物。在开始阶段,S原子很容易消除得到氮杂二烯41,随后立刻发生氮杂D-A反应,经过期望的endo过渡态,得到亚胺40及其对映体40’,经过异构化后,主要产物56及56‘经过氮杂Mannich反应得到57及57’,次要的40及40‘水解得到39及39’。
由上述机理可以看出,39及39‘的产率主要取决与亚胺及烯胺互变平衡,增加亚胺的比例自然可以提高39及39’的比例。于是,Nicolaou教授向其中加入卞胺,希望其高亲核性能够提高亚胺的消除。通过此方法,最终得到了60%的39及39‘,而只有痕量的57及57’,并且还有68%的醛49可以回收利用。
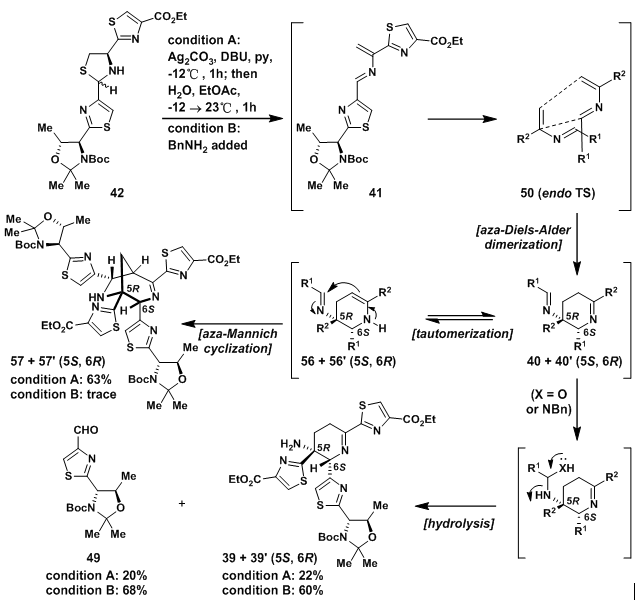
图9:Synthesis of dehydropiperidine system 39 via a biomimetic aza-D-A dimerization
在38的合成中,看似简单的胺合成反应也遇到了麻烦。39和39‘与59的反应并没有按照预期得到38及38‘,仔细分析发现,实际上得到了5元环状亚胺61和61’。虽然亚胺的亲核性不如初级胺,但是位阻效应在这起了更大的作用,于是初级胺先环化启动反应得到桥环过渡态60和60‘,随后发生环收缩,得到反应产物61和61’。
随后的实验中发现,位阻更小的亲电试剂可以正常的与大位阻的初级胺反应。于是设计酰氯64与其反应。L-甘氨酸的氨基先发生叠氮化反应[3],随后酰氯化得到64。在39和39‘与64中,以70%且无异构化产物的情况下得到期望产物65及65’。随后的反应中将乙酯置换为甲酯[4, 5],叠氮还原成氨基并用色谱柱分离两种构想,最后氨基经AllocCl保护得到38。
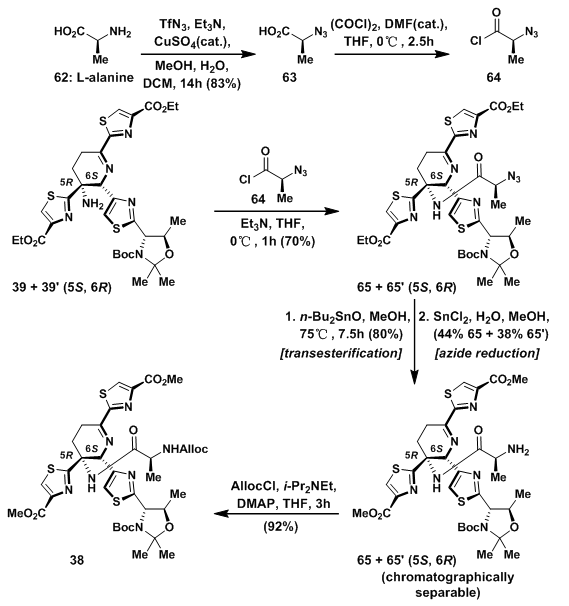
图10:Synthesis of dehydropiperidine fragment 38
2)其他碎片的合成
当归酸(Angelic acid)的sharpless不对称双羟化反应性并不好[6],改变至该条件后以90%的收率得到68(90:10dr)。邻位双羟基被缩酮化保护后,DIBAL-H还原得到初级醇69。初级醇先经过Dess-Martin氧化得到醛后,与苄基胺反应生成亚胺中间体70;随后用TMSCN终止反应,发生Strecker反应得到72,反应有良好的立体选择性主要是由5元环过渡态71中的位阻效应做到的;72发生氢解消除Boc保护基,随后在三乙胺及吡啶中与H2S发生反应,氰基转变成硫酰胺73;73发生另一分子的Hantzsch硫唑合成反应,随后N被保护得到74;74先发生酯交换得甲酯后,在TFA的作用下脱除保护基,然后羟基经TBSCl保护,得到甲酯75。

图11:Synthesis of thiazole fragment 75
碎片37是合成硫唑啉环不可缺少的一部分,L-苏氨酸衍生物76先经TFA脱保护,后发生叠氮化反应[3],得叠氮化物77;77发生水解反应得到羧酸78。L-苏氨酸衍生物79与甲酯80偶联形成二肽81,随后羟基保护得到82,再与Lawesson试剂反应得到硫酰胺83,脱除保护基得到初级胺84;胺84与78偶联得到85,随后脱除羟基保护得初级醇86;在DAST的协助下,86反应生成硫唑啉环,随后甲酯在锡试剂作用下得到羧酸87;羧酸7与胺75反应形成酰胺88,随后甲酯氢解得到碎片37。
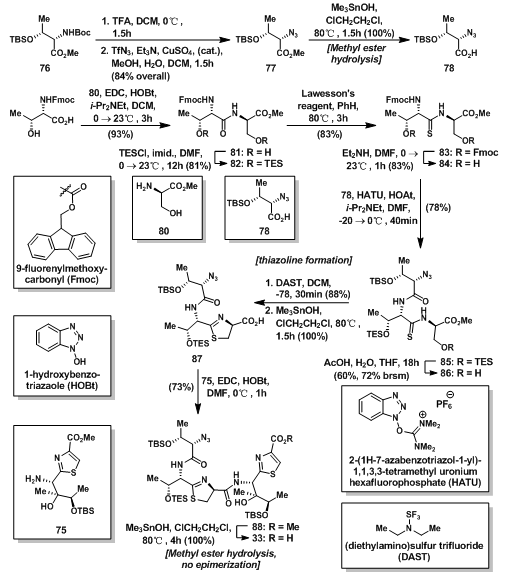
图12:Synthesis of thiazole-thiazoline fragment 37
含硒碎片34的合成是从L-甘氨酸衍生物89开始的,首先羧基经两步得到酰胺90,脱除Boc保护后的氨基与另一分子的89反应,形成另一酰胺键得到91,随后脱除保护基得34。
分子中另一部分含硒碎片35的部分结构也需从9出发合成,89与烯丙醇反应得到烯丙醇酯92,随后脱除Boc保护基的92与93反应,得到酰胺94,脱保护基后得胺95。
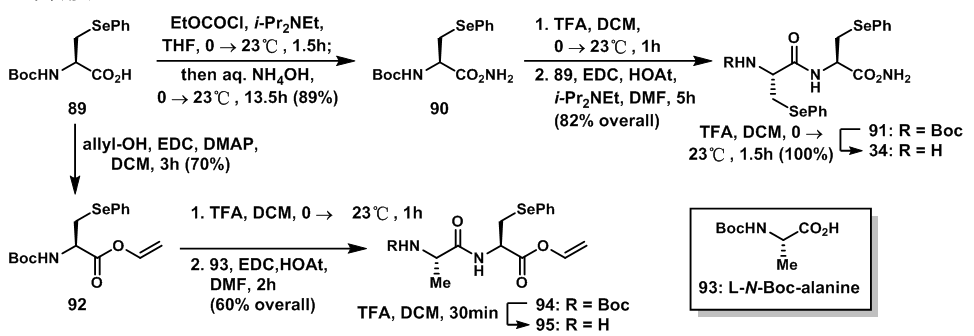
图13:Synthesis of dipeptide fragment 34 and 95
喹哪酸碎片35的合成是由2-羧基喹啉开始的,首先酯化得到甲酯后,氢化得吡啶98;98发生一分子的Fenton反应,得到甲基酮100;甲基酮100经CBS还原得到的羟基经TBS保护得103;然后经过3步的Boekelheide串联反应得到醇108[7](50),103先与m-CPBA反应得104,随后与TFAA反应得到过渡态105,去质子得过渡态106,然后发生Boekelheide反应得到107,再于碱性条件下水解得到醇108,其中106到107中的N-O键断裂的机理还不清楚,周环、阴离子、自由基都有可能;108最后与Burgess试剂反应,经过渡态109得到烯烃110。

图14:Synthesis of olefin 110
烯烃110的双键在发生不对称环氧化反应时遇到了一些意外的麻烦,最终选择Katsuki环氧反应得到希望的目标产物113(82%,87:13 dr);113发生自由机型的溴代反应得到114,随后消除得到α,β-环氧烯烃115。

图15:Synthesis of activated epoxide 115
115与116发生SN2反应得到醇之后用TBSOTf保护羟基得到117,随后发生酯水解得到羧酸118;羧酸118与FmOH发生酯化反应得119;Pd催化的还原消除反应及与95的偶联反应使119得到120,随后另一分子的Pd催化的消除反应得到羧酸35。
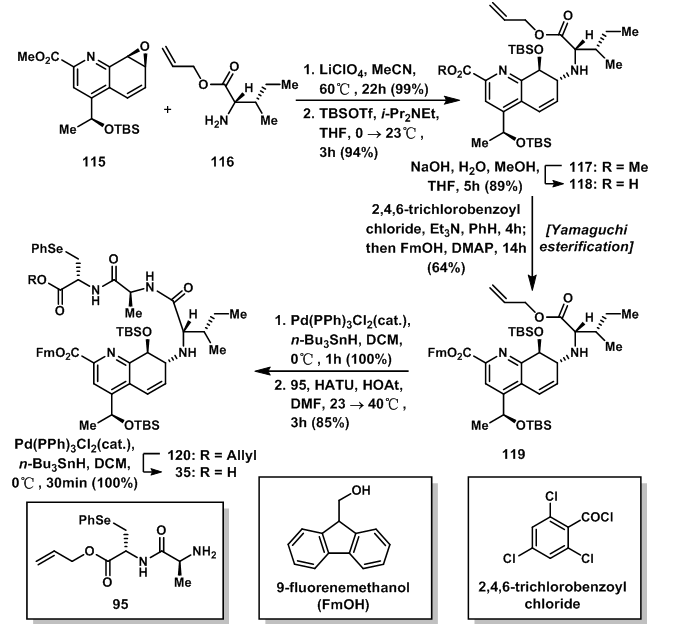
图16:Synthesis of quinaldic acid fragment 35
3)碎片偶联及thiostrepton全合成
虽然碎片已经全部拿到,但是thiostrepton全合成的难题仍然存在。设计的碎片偶联路线、各种保护基团的选择性在全合成中都有着很大的灵活度。在最终的路线中,选择了首先合成26元环碎片36,如下图所示,脱氢呱啶碎片38产生一个一级氨基,并与硫唑啉-硫唑碎片37偶联121;121中两个甲基酯的化学环境相似,区域选择性的氢解没有意外的难以实现,12氢解后得到52%的122及其另一个氢解产物122‘,12%的二酸及28%的回收原料121;122和122’经过Staudinger还原得到对应的氨基酸123及123‘;手工搭建的模型显示期望的123比123’更加适合发生大环酰胺化,于是在发生大环酰胺化反应后得到单一的产物124,随后氢解得到6元大环碎片36。
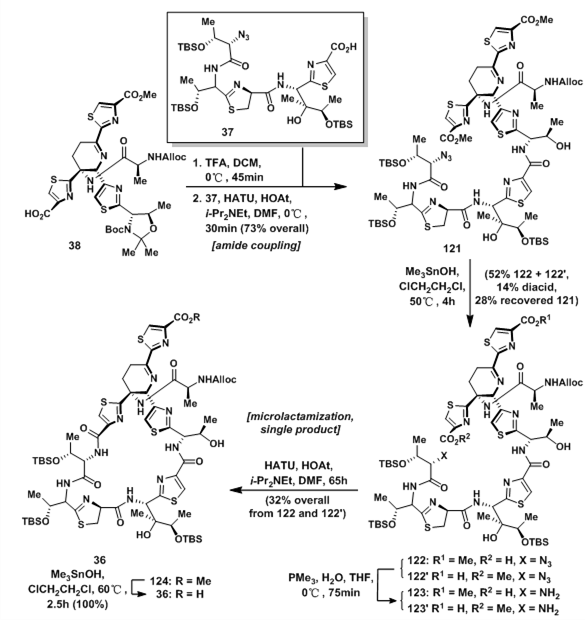
图17:Synthesis of macrocycle 36
大环羧酸36与二肽片段34发生偶联反应后得到125;随后脱除Alloc保护基后与35发生另一分子的酰胺偶联得到126,随后脱除Fm保护基得到羧酸127;羧酸127经Yamaguchi环酯化反应后得到33;脱氢呱啶是良好的Michael受体,并且对许多反应条件都非常敏感。Et2NH脱除Fm保护基和Pd催化剂都是相当温和的反应条件,但对脱氢呱啶却不能适用。叔丁基过氧酸使硒醚发生消除得到脱氢甘氨酸结构,随后用HF·py脱除中间体的Si保护基和脱水得到最终的天然产物thiostrepton。
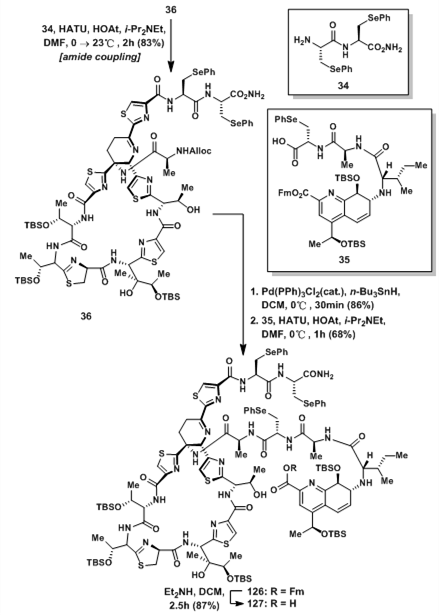
图18:Synthesis of advanced hydroxyl acid intermediated 127
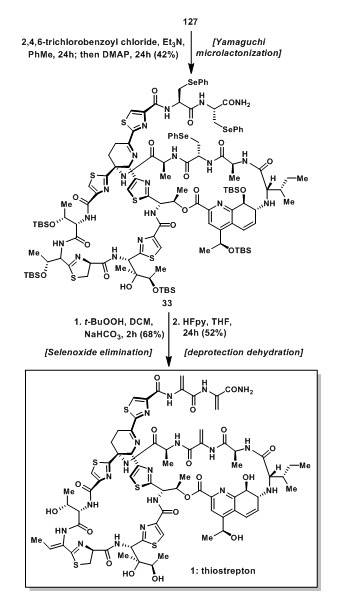
图19:Total synthesis of thiostrepton (1)
总结
仿生的氮杂D-A反应在脱氢呱啶的合成中起着至关重要的作用,并且在thiostrepton的合成中也有重要的作用。另外,作为温和的酯氢解反应,Me3SnOH是一种非常好用的试剂[8](45)。
参考文献
- For a review on the use of Lawesson’s reagent, see: T. Ozturk, E. Ertas, O. Mert, Chem. Rev., 2007, 107, 5210. DOI: 10.1021/cr040650b;
- a) C. W. Holzapfel, G. R. Pettit, J. Org. Chem., 1985, 50, 2323. DOI: 10.1021/jo00213a024. b) R. C. Kelly, I. Gebhard, N. Wicnienski, J. Org. Chem., 1986, 51, 4590. DOI: 10.1021/jo00374a019;
- a) B Alper, S-C Hung, C-H Wong, Tetrahedron Lett, 1996, 37, 6029. doi.org/10.1016 /0040-4039(96)01307-X; b) P. T. Nyffeler, C-H Liang, K. M. Koeller, C-H Wong, J. Am. Chem. Soc., 2002, 124, 10773. DOI: 10.1021/ja0264605;
- For a review on transesterifications, see: J. Otera, Chem. Rev., 1993, 93, 1449. DOI: 10.1021/cr00020a004;
- Giannis, P. Baumhof, R. Mazitschek, Angew. Chem. Int. Ed. 2001, 40, 3672.DOI: 10.1002/1521-3773(20011001)40:19;
- a) K. Barry Sharpless, W. Amberg, Y. L. Bennani, G. A. Crispino, J. Hartung, K. S. Jeong, H-L. Kwong, K. Morikawa, Z-M. Wang, J. Org. Chem., 1992, 57, 2768. DOI: 10.1021 /jo00036a003. b) For a review on asymmetric dihydroxylations, see: H. C. Kolb, M. S. VanNieuwenhze, K. B. Sharpless, Chem. Rev., 1994, 94, 2483. DOI: 10.1021/cr00032a009;
- a) V. Boekelheide, W. J. Linn, J. Am. Chem. Soc., 1954, 76, 1286. DOI: 10.1021/ja01634a026. b) S. Oae, S. Tamagaki, T. Negoro, S. Kozuka, Tetrahedron, 1970, 26, 4051. doi.org/10.1016/S0040-4020(01)93045-5. c) C. Fontenas, E. Bejan, H. AïtHaddou, G. G. A. Balavoine, Synth, Commun. 1995, 25, 629. doi.org/10.1080/00397919508011399;
- a) R. L.E. Furlán, E. G. Mata, O. A. Mascaretti, Tetrahedron Lett. 1996, 37, 5229. org/ 10.1016/0040-4039(96)01071-4. b) K. C. Nicolaou, A. A. Estrada, M. Zak, S. H. Lee, B. S. Safina, Angew. Chem. Int. Ed. 2005, 44, 1378. DOI: 10.1002/anie.200462207;
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!


















No comments yet.