
作者:石油醚
引言
近日, Pfizer的科学家聚焦于步骤1与步骤2的串联工艺开发,通过优化交叉偶联条件,简化后处理并实现p-TsOH·H₂O介导的直接串联脱保护,成功实现从2和3直接高效制备中间体5的无色谱、低金属残留、高收率的连续化操作。
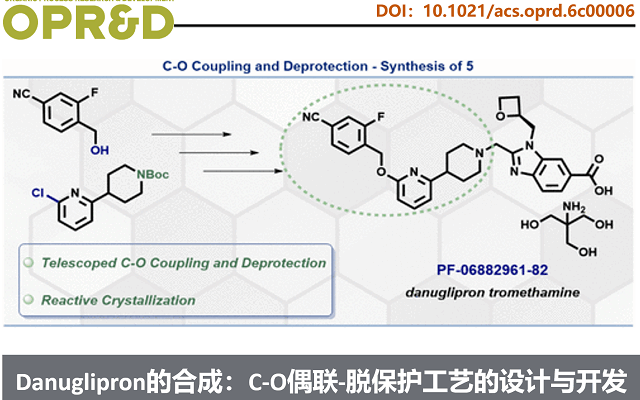
Synthesis of Danuglipron: Design and Development of a Robust Telescoped C–O Coupling─Deprotection Sequence.
Unjila Afrin, Aaron Baldwin, Arlene P. Bartolome, Nga M. Do, Andres R. Faria Quintero, David F. Fernández, Jonathan Fifer, Steven J. Fussell, Shanjun Huang, Gary R. Jolin, Md Kamrul Hasan Khan, Melissa Lee, Taegyo Lee, Laura McGivern, Giselle P. Reyes, Rachel Ruest, Adam Scott, Ursula Sheridan, Steven J. R. Twiddle, Angela L. A. Puchlopek-Dermenci, Chase Anthony Salazar, Sergei Tcyrulnikov, Gerald A. Weisenburger, Bianca Williams, and Yexenia Nieves-Quinones*
Org. Process Res. Dev. 2026. DOI : 10.1021/acs.oprd.6c00006
正文
活Danuglipron tromethamine (PF-06882961-82, 1) 是辉瑞开发的口服非肽类GLP-1受体激动剂,用于2型糖尿病和慢性体重管理。与现有获批的肽类GLP-1R激动剂(均需皮下注射)不同,Danuglipron可每日口服一次;司美格鲁肽是目前唯一兼具注射与口服剂型的GLP-1R激动剂。其药理作用包括葡萄糖依赖性促胰岛素分泌、抑胰高血糖素释放、延缓胃排空、增强饱腹感及减少摄食。在停止开发之前,Danuglipron(1)是少数几种在研的非肽类 GLP-1R 激动剂候选药物之一。
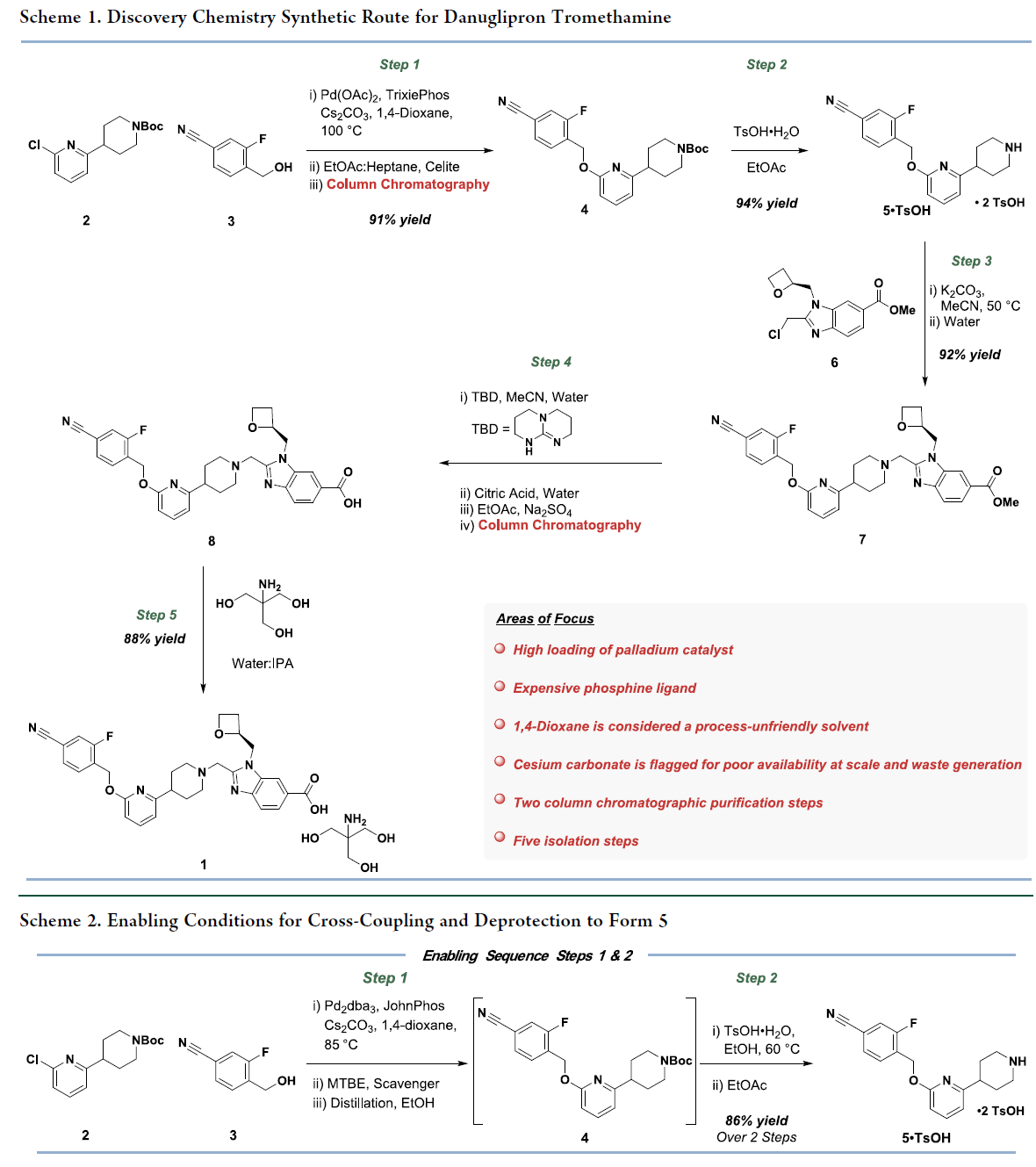
Danuglipron的合成方法,如scheme 1所示。即2与3经钯催化C–O偶联获得4,4脱保护得二对甲苯磺酸盐5。随后,5在碱性条件下与含氧杂环丁烷的氯甲基苯并咪唑6发生烷基化,生成酯7。最后,7经TBD介导水解得中间体8,再与三羟甲基氨基甲烷(tromethamine)成盐,得到Danuglipron(1)。该路线虽成功支持临床前供药,但存在明显工艺瓶颈:步骤1钯用量高(5 mol%),且全程需两次柱层析纯化。鉴于danuglipron具有重大临床与商业化潜力,亟需建立低成本、稳健、高效、可持续的商业化生产工艺。基于此,Pfizer的科学家聚焦于步骤1与步骤2的串联工艺开发——即从2和3直接高效制备中间体5,实现无色谱、低金属残留、高收率的连续化操作。
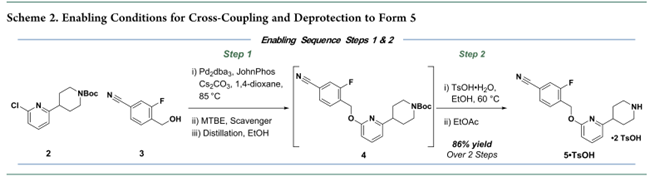
首先,对原路线的步骤1和2进行了工艺优化(Scheme 2)。关键改进包括:以更廉价、易得的JohnPhos配体替代Trixiephos,将钯用量从5 mol%降至2 mol%;并实现步骤1与后续脱保护的直接串联,取消中间色谱纯化。但原有C–O偶联仍存在显著缺陷:采用高风险溶剂1,4-二氧六环和高成本、高密度、高废重的碳酸铯(Cs₂CO₃)作碱。该组合导致钯残留高,需额外硅藻土过滤、硅胶捕获及乙醇溶剂置换,大幅增加操作步骤、成本与时长。脱保护环节使用对甲苯磺酸,经结晶得双对甲苯磺酸盐5,但因晶体细小,形成稠厚浆料,过滤困难、速率慢。基于此,作者确定了工艺优化和改进工作主要围绕五个关键目标展开:(1)降低钯和配体的成本;(2)确定一种更易获取的长期生产用碱;(3)用更可持续的溶剂替代 1,4-二氧六环;(4)通过减少单元操作和缩短循环时间来简化工艺;(5)评估中间体 5 的潜在盐型。
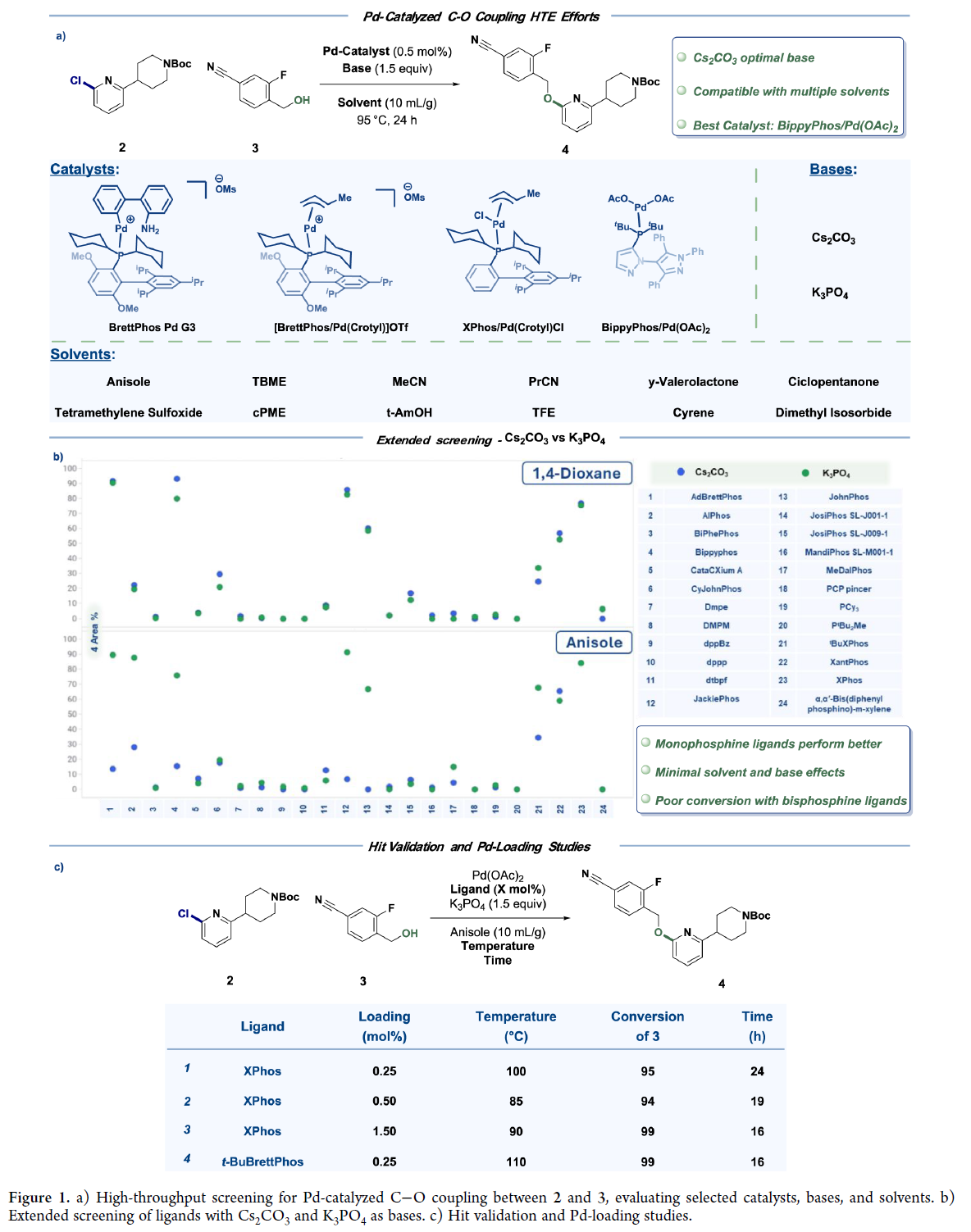
初期尝试以铜替代钯催化剂,旨在降低成本并提升可及性。高通量实验(HTE)筛选出DPEO和BNMO作为潜在铜配体,但优化后产率仅65–69%,且反应对氧气敏感、杂质多;与后续酸脱保护串联时,所得5•TsOH纯度低,需重结晶。综合评估表明铜催化不具稳健性,遂转向钯催化路线。即在钯催化的条件下,作何筛选首先考察12种溶剂、2种碱及多种催化剂(Figure 1a)。结果显示:Cs₂CO₃在多数溶剂中优于K₃PO₄,但其高成本、高密度及废固等问题制约放大。进一步针对24种市售膦配体(Figure 1b)的筛选发现,富电子大位阻单膦(JackiePhos、BippyPhos、AdBrettPhos、AlPhos、XPhos等)可实现2到4的高效转化;而双/三烷基膦普遍转化不完全。其中,关键发现是:溶剂与碱对转化率影响微弱,1,4-二氧六环和Cs₂CO₃可被苯甲醚和K₃PO₄替代。苯甲醚体系重现性好,并支持钯载量显著降低(Figure 1c)。
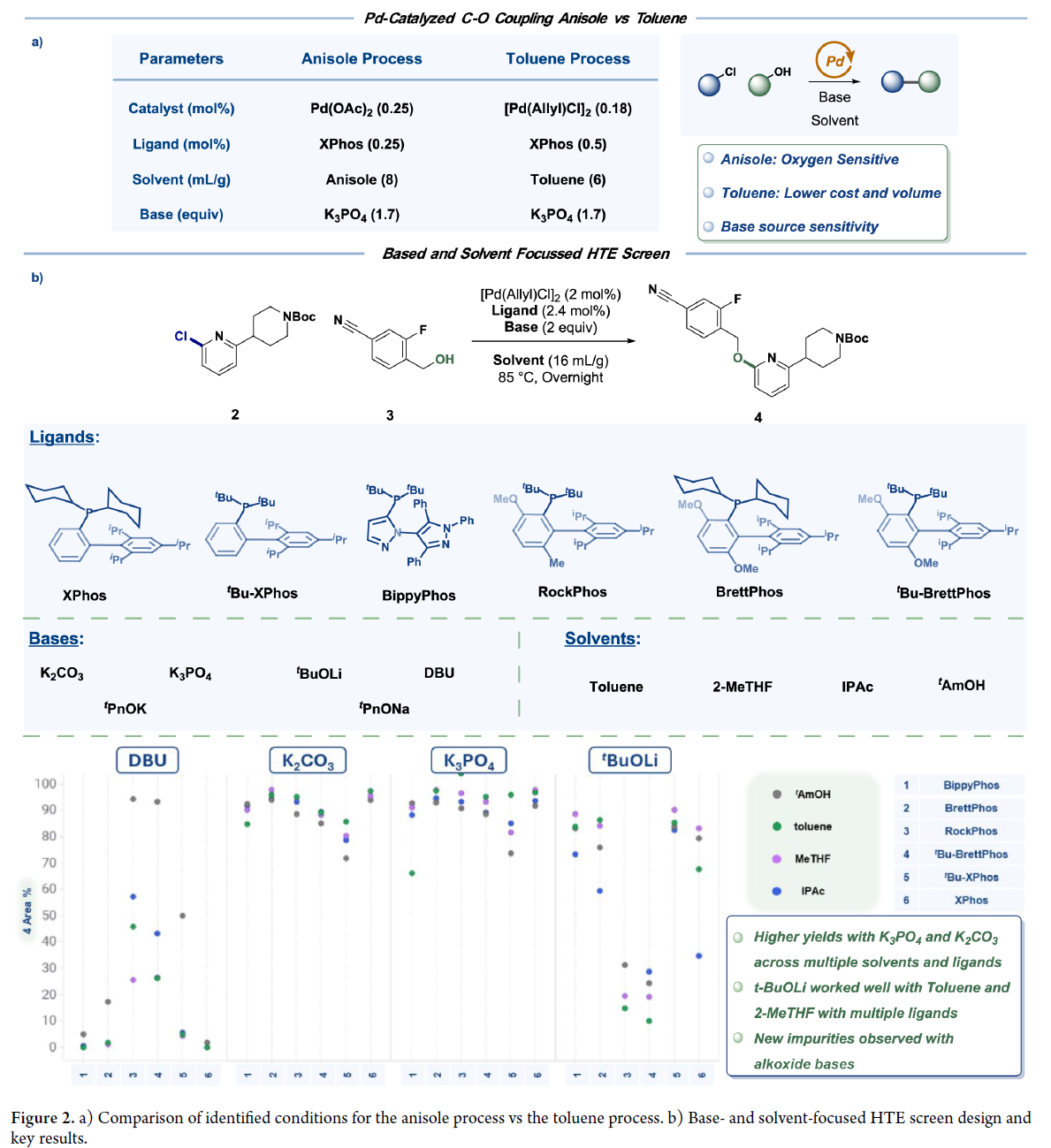
经综合评估配体/催化剂的性能、成本与可及性,选定XPhos/Pd(OAc)₂为最优组合。优化得放大工艺:XPhos(0.25 mol%)、Pd(OAc)₂(0.25 mol%)、K₃PO₄(1.7 equiv)在苯甲醚(10 mL/g)中100 °C反应,成功交付16.4 kg 5•TsOH·H₂O(收率82%)。但苯甲醚成本高、供应受限,推动溶剂替换研究。甲苯:醇混合体系因物理性质差、杂质高被排除;单一甲苯体系表现优异。进一步筛选确认:(allylPdCl)₂/XPhos组合在甲苯中反应更快、杂质更低、收率更高。对比评估显示甲苯工艺稳健性优于苯甲醚(Figure 2a)。K₃PO₄性能高度依赖供应商及物态(无水vs.一水合物):粉末状无水K₃PO₄(小粒径)反应快;一水合物因颗粒粗大导致反应停滞。为此,转向筛选更易控粒径或溶解性更好的碱。
Figure 2b筛选显示:K₂CO₃(78–98%收率)和K₃PO₄(66–98%)在多数溶剂中表现优异;tBuOLi和DBU虽有效,但需大幅变更工艺,未深入开发。验证表明:甲苯中XPhos/K₂CO₃体系收率达84%,且K₂CO₃易获指定目数产品,可消除K₃PO₄批次差异,工艺变更最小。最终确立甲苯体系最优条件:(allylPdCl)₂(0.18 mol%)、XPhos(0.50 mol%)、K₂CO₃(1.7 equiv)。小试(≤5 g)可过夜完全转化;但中试放大时出现反应终点波动及升温速率敏感等问题,提示工艺已逼近失败边缘,存在批间失败风险。为此启动动力学建模,以解析变异性根源,定义具备充分安全裕度的稳健操作窗口。
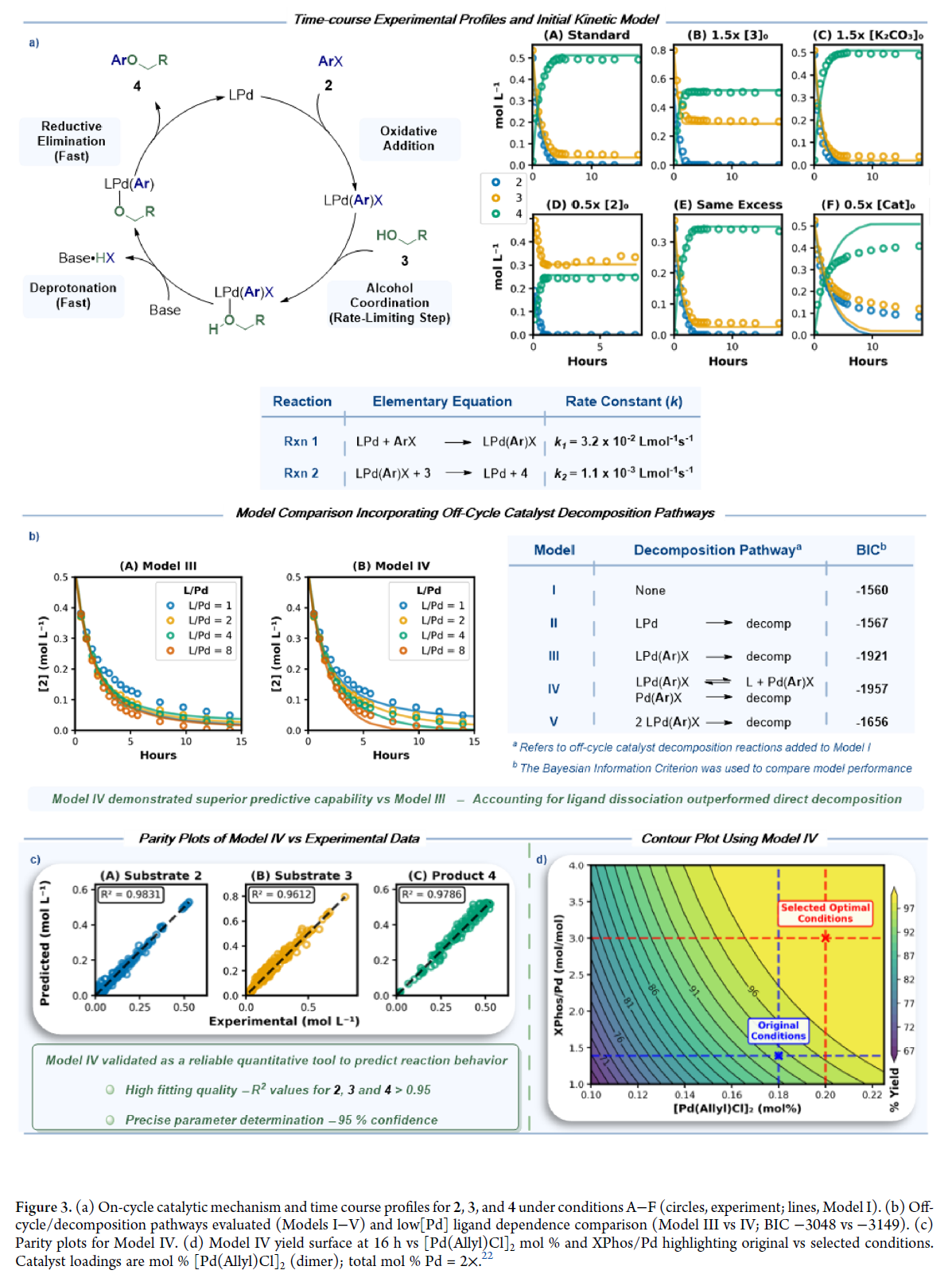
鉴于上述原因,作者通过设计实验来定量研究反应动力学。即在100°C、5 mL甲苯中,系统改变反应条件,采集化合物2、3、4的时间浓度数据。取样采用ReactAll平台20,UPLC-MS定量,以2,2′-二甲基联苯为内标。共设计六组实验(Figure 3a): (A) 标准条件:[2]₀ = 0.51 M,[3]₀ = 0.54 M(1.05 eq),[K₂CO₃]₀ = 0.87 M(1.7 eq),[[Pd(allyl)Cl]₂]₀ = 1.38 mM(0.54 mol% Pd),[XPhos]₀ = 3.83 mM(0.75 mol%); (B) [3]₀ ×1.5; (C) [K₂CO₃]₀ ×1.5;(D) [2]₀ ×0.5; (E) [2]₀、[3]₀、[K₂CO₃]₀ 各降0.16 M; (F) [[Pd(allyl)Cl]₂]₀ 和 [XPhos]₀ 均×0.5——作为低负载压力测试,探查失效边界;A–E则围绕高负载稳健区展开参数空间。
基于Buchwald提出的C–O偶联机制,作者构建并简化多个动力学模型。模型I(Figure 3a)假设氧化加成快速、醇配位为速率决定步,最佳拟合A–E数据。40°C反应1 h后的³¹P NMR显示53.2 ppm特征峰,证实氧化加成中间体为催化休眠态²¹,支持该模型。但模型I在条件F下显著偏离——初速率吻合好,后期偏差增大,表明低负载时催化剂更易失活。为此,我们在模型I基础上引入失活路径(Figure 3b): II:Pd(0)失活; III:氧化加成中间体直接分解; IV:先发生配体解离,再失活; V:氧化加成中间体二聚失活。BIC评估显示,III(−1921)和IV(−1957)远优于I(−1560),且二者接近,说明失活主要经氧化加成中间体,无论是否经配体解离。 进一步为区分III与IV,在[Pd]₀ = 0.14 mol%下系统调节XPhos/Pd比(1–8)。结果表明:IV全面优于III——尤其在L/Pd = 1时,III严重高估底物消耗;在高L/Pd下亦出现偏差;而IV在全部比例下均精准复现时间进程。BIC进一步确认:IV(−3149)> III(−3048)。该结果证实失活源于配体解离平衡,解离后裸露的Pd(II)物种更易降解——这也解释了为何低配体浓度加剧失活。然而,模型IV在全数据集上验证效果优异(Figure 3c):对2、3、4的预测R²分别为0.9829、0.9592、0.9779;所有速率常数95%置信区间宽度<35%,参数高度可靠。
基于该模型开展工艺优化(图3d):在[Pd]₀ = 0.1–0.225 mol%、XPhos/Pd = 1–4范围内模拟16 h产率。原条件(0.18 mol% Pd,L/Pd = 1.39)预测产率93.9%,已临近失活区;最优条件为0.20 mol% Pd + L/Pd = 3.0,预测产率99.7%。该调整将工艺移出失效边缘,显著提升鲁棒性与安全裕度。核心操作启示:在低钯负载下,提高XPhos/Pd比可有效抑制配体解离引发的失活,维持高活性双配位Pd(II)物种浓度。实践准则即——极低Pd用量必须匹配高配体当量,以保障催化体系长期稳定高效运行。
由于模型IV确定了最优交叉偶联条件,显著提升工艺安全裕度。后续工作聚焦三方面:步骤1的后处理优化、脱保护反应开发,以及5以TsOH盐形式的串联分离。 C−O偶联后处理:原方案需两次过滤(先除碱/盐,再除金属),放大时因过滤慢、操作多成为瓶颈。改用水洗替代——采用乙醇/乙酸乙酯共溶助溶,实现快速、清晰的两相分离;有机相直接用于p-TsOH·H₂O介导的脱保护。 甲苯体系中,以MEK替代乙酸乙酯,保持水/乙醇用量不变,同样获得良好相分离和溶液收率。 但5•TsOH中钯含量波动,且步骤2结晶重现性差。 为强化钯清除,系统筛选30种水溶性金属清除剂(室温及70°C液-液萃取)。结果表明:升温显著提升效率;L-半胱氨酸与N-乙酰-L-半胱氨酸效果最佳(~70%钯去除)。固体清除剂(碳基、硅基、硫脲类)无效,故弃用。 同步简化后处理:原三溶剂体系(甲苯/乙酸乙酯/乙醇)旨在抑制乳化与絮状层。我们推测步骤2结晶异常源于有机相含水量波动——可能受混合后静置时间影响。单次水洗即有效抑制絮状层,相分离清晰、重现性好。

接下来,作者对5的盐型进行了筛选,结果表明:其他酸(HCl、MsOH、草酸、各类磺酸)所得盐均结晶性差、重现性低;而双对甲苯磺酸盐(5•TsOH)结晶稳定、批次一致,且虽含约47%对甲苯磺酸根,但综合性能最优,故选定为最终盐型。脱保护条件经系统优化:考察溶剂比例(甲苯/MEK/乙醇)及p-TsOH·H₂O当量对结晶质量、浆料稠度和产物纯度的影响。结合溶解度与温度依赖性研究,确定最优条件为:p-TsOH·H₂O 2.5 eq,甲苯 7 mL/g,MEK 3 mL/g,乙醇 2 mL/g。该体系实现5•TsOH一水合物的直接反应结晶,无需晶种诱导。
前期引入水相后处理(L-半胱氨酸水洗)已消除过滤瓶颈,并显著提升5•TsOH质量。PXRD与偏光显微镜(PLM)分析证实:水相环境主导晶型——仅在水存在下稳定获得一水合物;非水体系则得针状无水物(Figure 4)。但改用含水金属清除剂后,成核过快,导致晶体细小、夹杂杂质、晶型混杂(一水/半水/无水共存)。 为此,在脱保护中精准补加3 eq水:有效调控成核速率,得到大而规整的板状一水合物晶体,兼具高过滤速率与优异杂质清除能力。
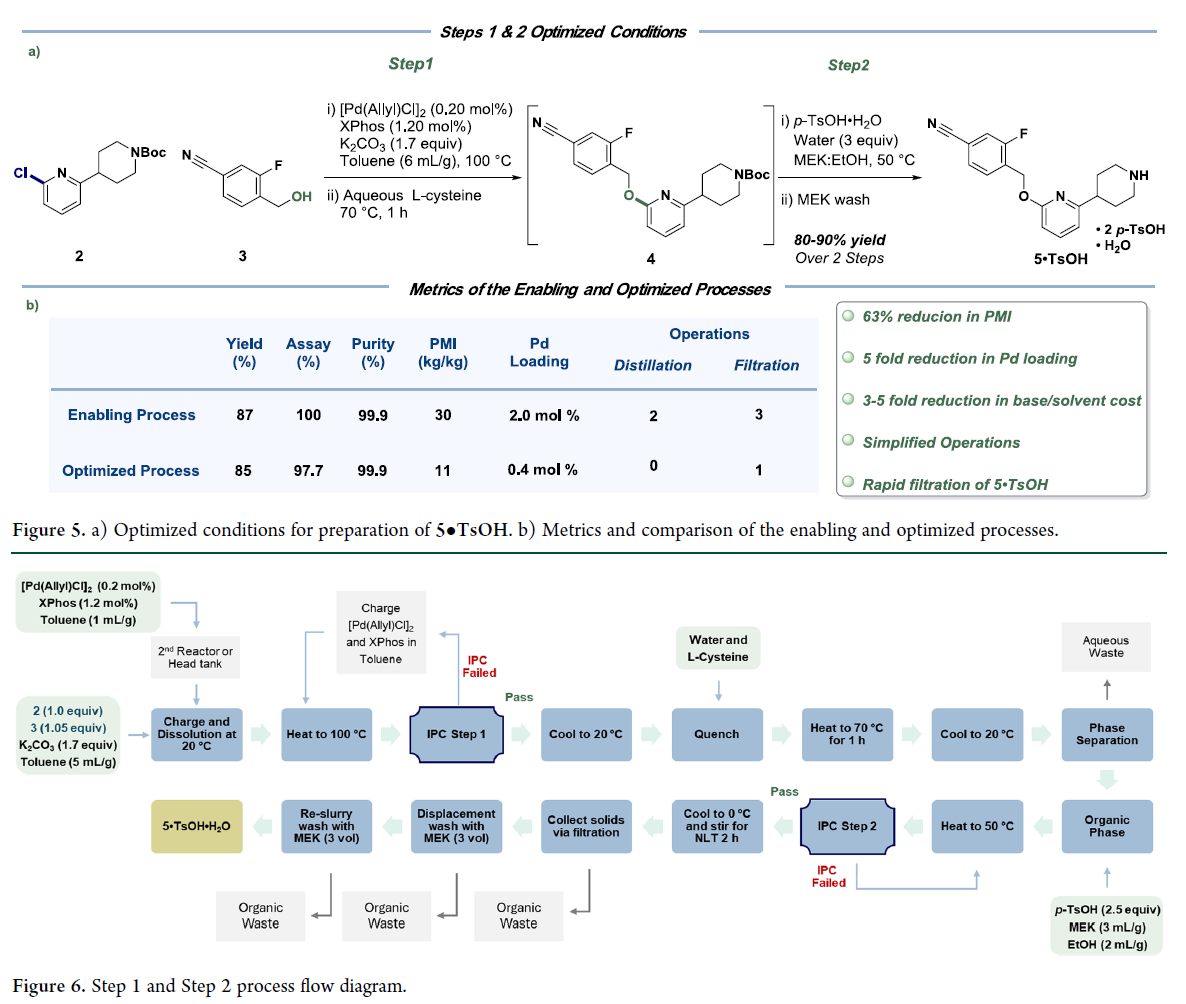
最终工艺(Figure 5a):甲苯中Pd(allyl)Cl₂/XPhos/K₂CO₃催化C–O偶联 → L-半胱氨酸水溶液热洗(除Pd)→ 有机相加p-TsOH·H₂O于50°C反应结晶 → 直接过滤得高纯5•TsOH一水合物。与启用工艺对比(Figure 5b):钯用量从2.0 mol%降至0.4 mol%,碱与溶剂消耗降低3–5倍,PMI由30 kg/kg大幅降至11 kg/kg(−63%);全程取消蒸馏与多次过滤,操作步骤精简,质量稳定。该优化工艺兼具鲁棒性、经济性与绿色可持续性,适用于规模化生产。
结论
Pfizer的科学家聚焦于步骤1与步骤2的串联工艺开发,通过优化交叉偶联条件,简化后处理并实现p-TsOH·H₂O介导的直接串联脱保护,成功实现了从2和3直接高效制备中间体5的无色谱、低金属残留、高收率的连续化操作。Figure 6所示两步串联工艺仅需一次相分离和一次结晶隔离,即得高纯度5•TsOH一水合物;累计交付约1吨该中间体。 四批生产平均分离收率80%。新工艺减少单元操作,采用廉价易得的钯源、配体和无机碱,并改用更适宜放大的溶剂,使PMI降低63%,显著提升稳健性、可放大性,同时降低操作复杂度与成本。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/

















No comments yet.