
本文作者:石油醚
导读:
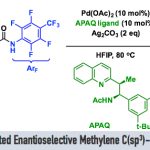
近日,匹兹堡大学的王亦鸣课题组在J. Am. Chem. Soc. 中发表论文,报道铱催化的通过碳氢键活化过程进行的炔丙位烯丙基化反应。该反应能通过良好的收率和立体选择性得到不对称的1,5-烯炔产物,具有广泛的底物范围,并且能通过配体的选择得到不同的非对映异构体。这一反应进一步发展了金属配位活化炔丙基位质子的策略,拓宽了铱-手性亚磷酰胺催化体系的应用,为手性催化提供了新的见解。
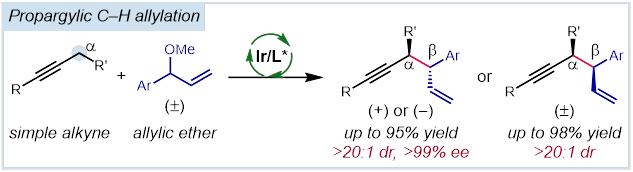
“Enantioselective and Diastereodivergent Allylation of Propargylic C–H Bonds.
Jin Zhu, Yidong Wang, Aaron D. Charlack, and Yi-Ming Wang*.
J. Am. Chem. Soc. 2022, 144, 15480–15487. doi : 10.1021/jacs.2c07297”
正文:
近年来,碳氢键的不对称官能团化取得了长足的发展,已经成为了一种利用简单的起始材料合成含有立体中心的产物的高效的方法。手性炔烃广泛存在于天然产物、药物和功能性分子中,一直是有机化学不对称合成的研究热点。由于简单炔烃易于制备,因此在简单炔烃的α位对映选择性地构建碳碳键或碳杂原子键从而得到手性炔烃就成为了重要的研究目标。以往合成手性炔烃的工作主要依赖于使用预官能团化的炔烃进行不对称炔丙基化,或利用乙炔基金属前体对亲电试剂进行对映选择性亲核加成。而炔丙位碳氢键不对称官能团化的方法则较为局限(Figure 1A),目前已报道的过程主要利用了卡宾或氮宾对于碳氢键的插入反应。加州大学圣巴巴拉分校的张立明教授团队另辟蹊径,利用手性金催化剂,首次利用炔烃α位质子活化的策略以高的立体选择性实现了分子内的炔丙位官能团化。由于不对称炔丙位碳氢键活化还能够在炔烃的α- 和 β- 位置同时建立立体化学中心,因此这一策略具有很高的应用价值(Figure 1B)。
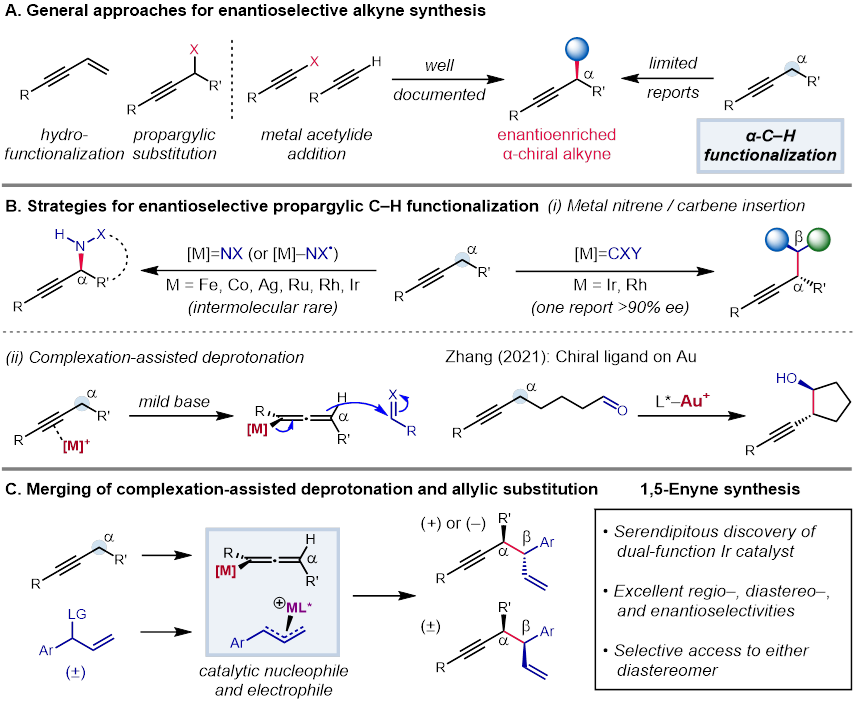
Figure 1. Asymmetric propargylic functionalization for 1,5-enyne synthesis
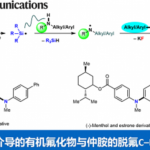
作者课题组在前期的研究中,发展了一系列取代茂环的羰基铁配合物,利用了金属配位不饱和键并活化α位质子的策略实现了简单炔烃、烯烃2以及联烯3的惰性碳氢键活化。受到这些研究成果的启发,作者希望通过利用在这一过程中生成的具有亲核性的铁中间体与含有不对称中心的π-烯丙基金属亲电试剂进行反应,从而得到具有立体选择性的产物。在初步的当量反应预实验中,由炔烃合成的联烯基铁中间体和π-烯丙基铱中间体之间的反应成功地生成了预期的1,5-烯炔产物(Figure 2A)。随后的协同催化实验也获得了相同的产物,验证了作者对反应过程的设计。然而,对照实验显示这一反应并不需要铁催化剂的参与(Figure 2B)。作者因此设想在这一过程中,铱催化剂不仅促进了π-烯丙基亲电试剂的生成,另外还取代了铁催化剂的作用,同时活化了炔烃底物,成为·了一种具有双功能的催化剂(Figure 1C)。与此同时,由于手性配体的控制,这一反应能够得到不对称的1,5-烯炔产物。
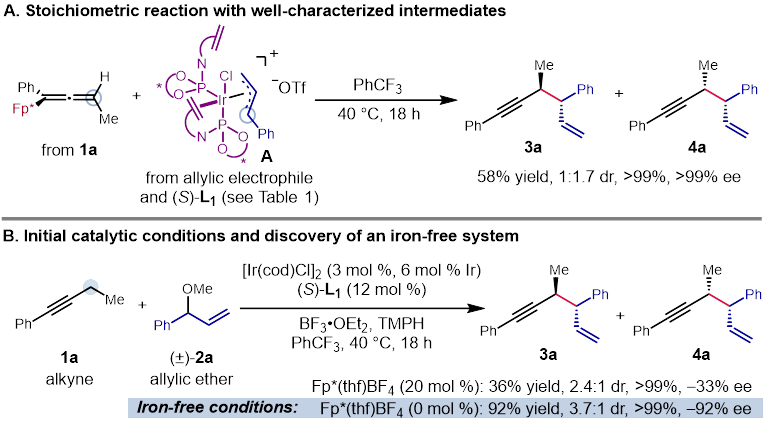
Figure 2. Preliminary results for propargylic C−H allylation
作者在对于反应条件进行一系列筛选后,以非常好的立体选择性得到了具有手性的反式烯炔产物。更有趣的是,利用非手性的亚磷酰胺类配体能以非常好的选择性得到消旋的顺式烯炔产物,而对于催化剂的进一步的改变大大提高了这一非对映异构体的收率。在最优的条件下,作者对这一立体发散反应的底物范围进行了探索。研究表明,带有给电子或吸电子取代基的1-芳基-1-丁炔能够有效地转化为相应的 1,5-烯炔,对于包括噻吩、呋喃、苯并呋喃和邻苯二甲酰亚胺在内的杂环也有非常好的官能团兼容性。当炔烃底物中的乙基被更长链的烷基取代时,需要加入更多的催化剂才能得到满意的结果。共轭烯炔以及芳基丙炔衍生物同样取得了成功的结果。最后,作者还测试了二烷基取代的炔烃的反应活性,虽然能够分离出单一的对映异构体,然而遗憾的是收率较低,显示了这一方法的局限性。此外,作者以5 mmol的规模合成了具有对甲苯磺酸酯的长链烷基取代基的产物,能通过对反应结束后的混合物直接重结晶的方法以73%的收率得到单一的立体异构体(Figure 3)。另一方面,具有吸电子取代基的芳基烯丙基醚也能够以高的收率和立体选择性得到相应的1,5-烯炔产物(Figure 4)。不仅如此,作者挑选了一些有代表性的炔烃和烯丙基醚底物,探究在外消旋配体存在的条件下生成顺式非对映异构体的反应活性。在这些底物中,均以非常好的选择性和相当的产率得到了立体结构反转的产物(Figure 5)。
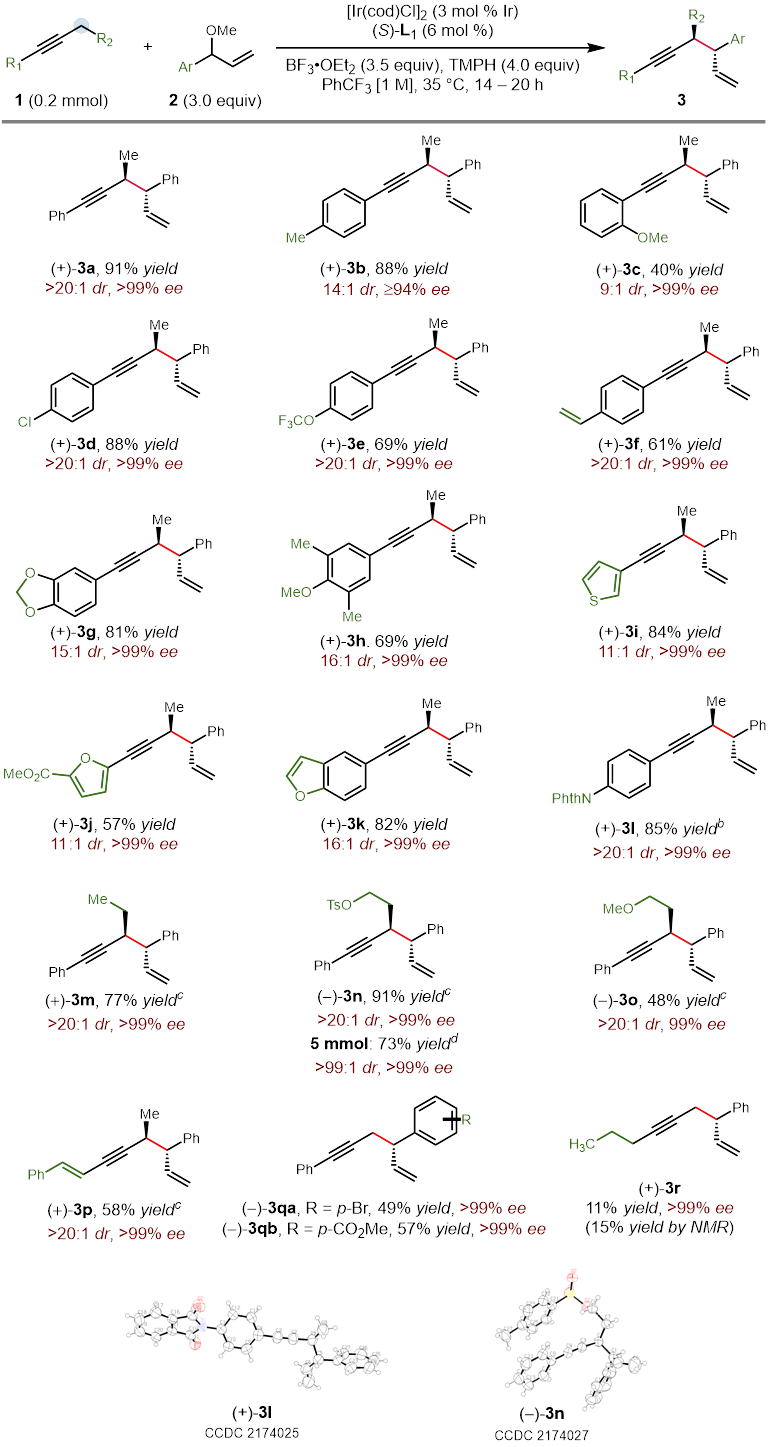
Figure 3. Substrate scope for the alkyne coupling partner
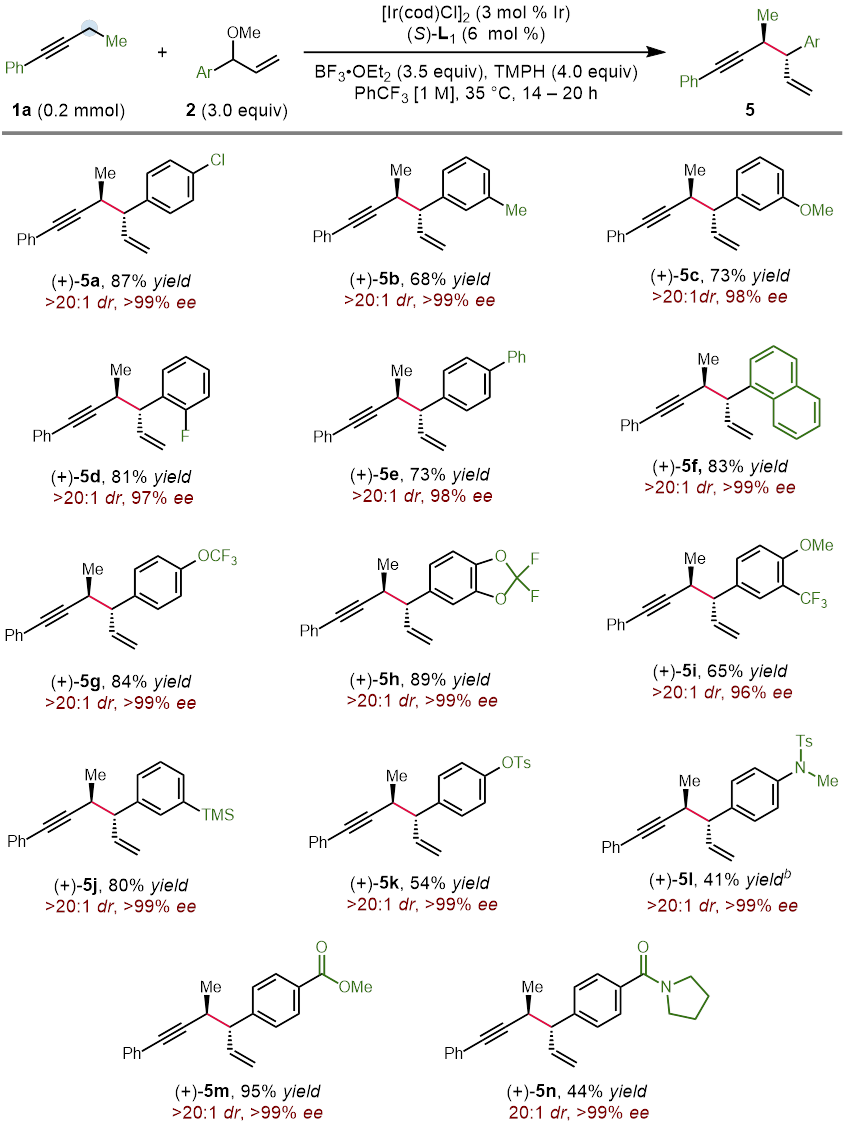
Figure 4. Substrate scope for the allylic ether coupling partner
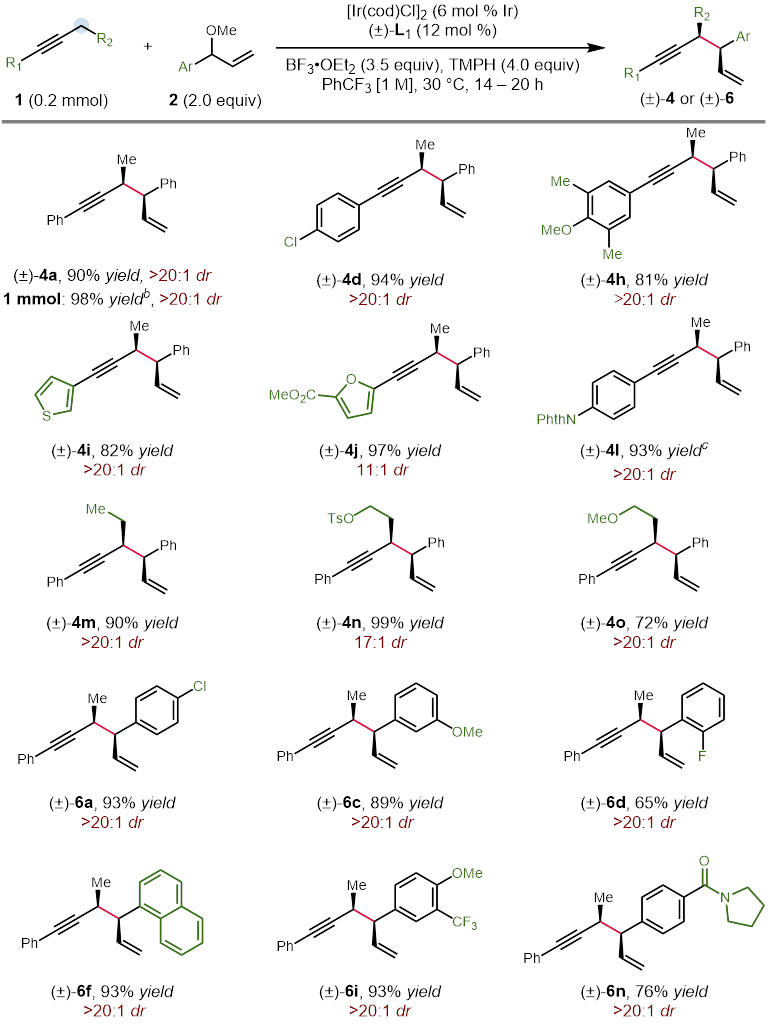
Figure 5. Diastereoselective synthesis of the syn-isomer
随后,作者进行了一系列的实验,以深入了解反应的机理(Figure 6)。对于反应过程的监测提示了诱导期的存在,而普通炔烃和氘代炔烃之间的反应速率差异则表明了显著的动力学同位素效应 (KIE)。在分子间竞争的反应条件下,测得了kH/kD 为 4.5的动力学同位素效应。综合起来,这些数据表明了在炔丙基位置的碳氢键活化是这一反应的限速步骤(Figure 6A)。
为了探究在这一转化中的立体控制过程,作者研究了配体的对映体过量值与产物中立体异构体组成之间的关系(Figure 6B)。在反应中,配体与产物之间的对映体过量的值存在着非线性关系,这在反式的烯炔产物中尤为明显,表明在反应过程中的金属中心上有两个手性配体进行配位。
烯丙基醚底物的光学纯度也对最终产物的立体选择性也有所影响(Figure 6C)。当(R)构型烯丙基醚参与反应时,产物的非对映选择性与使用(S)构型烯丙基醚用作起始原料时观察到的产物的非对映异构体比例存在很大差异,反应过程中也能观察到活性更低的(R)构型烯丙基醚的富集。这些实验表明了铱配位的烯丙基醚中甲氧基的离去很有可能发在炔烃配位以及碳氢键活化之后,进一步揭示了决定反应的非对映选择性的步骤的机理。
在这些结果的基础上,作者提出了他们对反应机理的猜测(Figure 6D)。通过对反应混合物的31P核磁共振检测,作者发现π-烯丙基铱配合物A在反应过程中一直是主要的含磷物质,结合这一配合物也是该反应的有效催化剂的实验结果,由于有诱导期的存在,作者提出了这一18电子络合物是反应中活性催化剂的来源的假设。另外,在反应中加入催化量的卤素离子以及事先利用银离子结合催化剂中氯离子的实验结果从侧面验证了作者的另一个猜想,即在反应过程中氯离子会从铱金属中心上解离,在失去氯离子的情况下形成的16电子烯丙基醚配合物I能够结合炔烃得到阳离子中间体II,再经过碱的去质子化得到中间体III。随后经过对烯丙基醚的氧化加成和在金属中心发生的还原反应顺理成章地得到了产物,而烯丙基醚与活性中间体V则能通过配位得到I,从而关闭了催化循环。
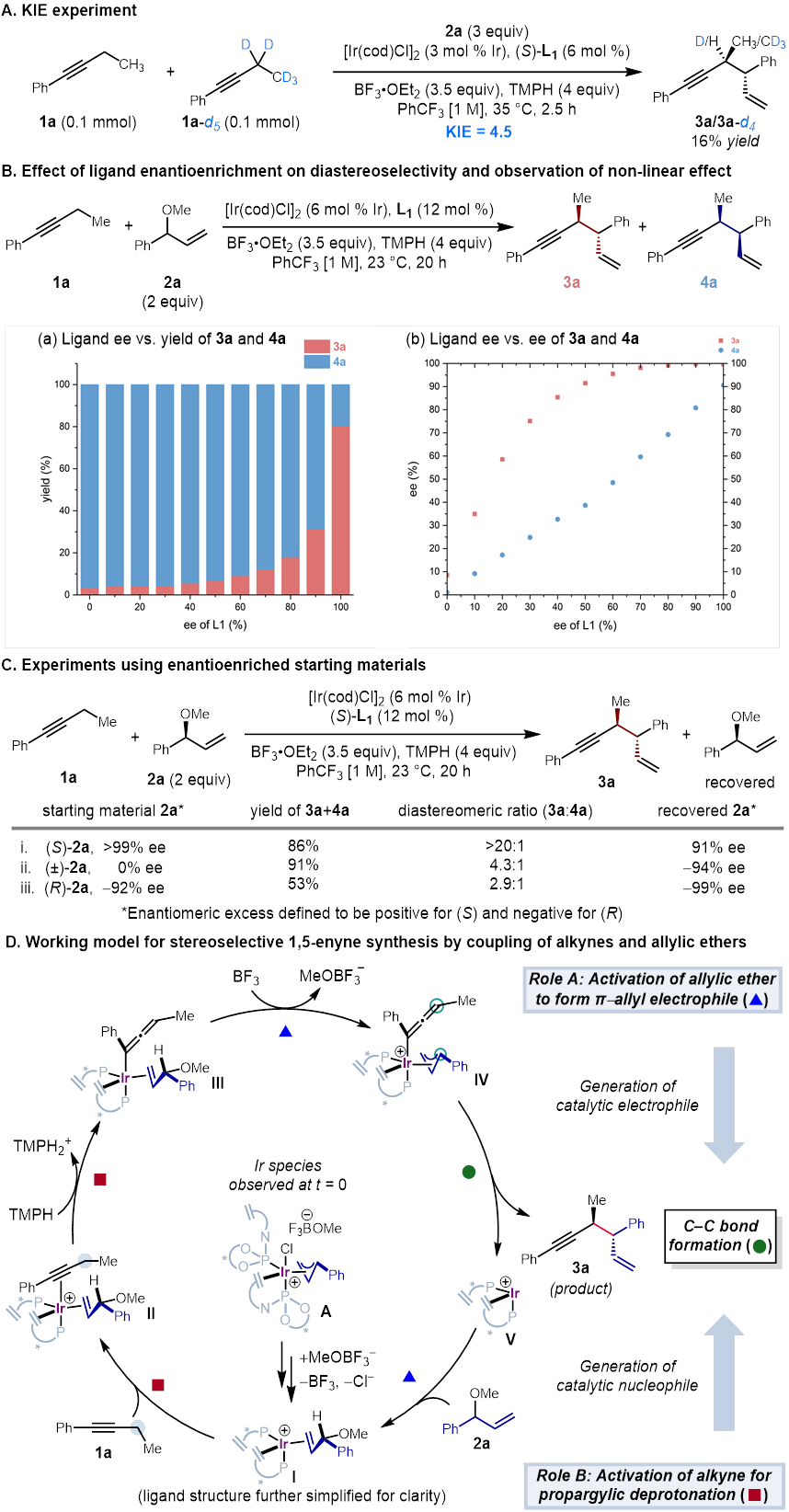
Figure 6. Selected mechanistic studies and proposed catalytic cycle
总之,作者描述了铱催化炔丙位碳氢键烯丙基化立体选择性地生成烯炔产物的过程,在这一过程中能够通过配体的改变得到立体发散的产物。作者同时提出了对于铱-亚磷酰胺催化体系全新的理解,后续也会在这一方面进行更为详细的机理研究。匹兹堡大学博士研究生朱锦为本论文的第一作者,王亦鸣教授为通讯作者,博士后研究员王以栋(现扬州大学校特聘教授)与研究生Aaron Charlack参加工作。
(王亦鸣教授供稿)
参考文献:
- [1] Li, T.; Cheng, X.; Qian, P.; Zhang, L. Gold-Catalysed Asymmetric Net Addition of Unactivated Propargylic C-H Bonds to Tethered Aldehydes. Nat. Catal. 2021, 4, 164–171.
- [2] Wang, Y.; Zhu, J.; Durham, A. C.; Lindberg, H.; Wang, Y.-M. α-C–H Functionalization of π-Bonds Using Iron Complexes: Catalytic Hydroxyalkylation of Alkynes and Alkenes. J. Am. Chem. Soc. 2019, 141, 19594−19599.
- [3] Wang, Y.; Scrivener, S. G.; Zuo, X.-D.; Wang, R.; Palermo, P. N.; Murphy, E.; Durham, A. C.; Wang, Y.-M. Iron-Catalyzed Contrasteric Functionalization of Allenic C(sp2)–H Bonds: Synthesis of α-Aminoalkyl 1,1-Disubstituted Allenes J. Am. Chem. Soc. 2021, 143, 14998–15004.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载

















No comments yet.