
本文作者:杉杉
导读
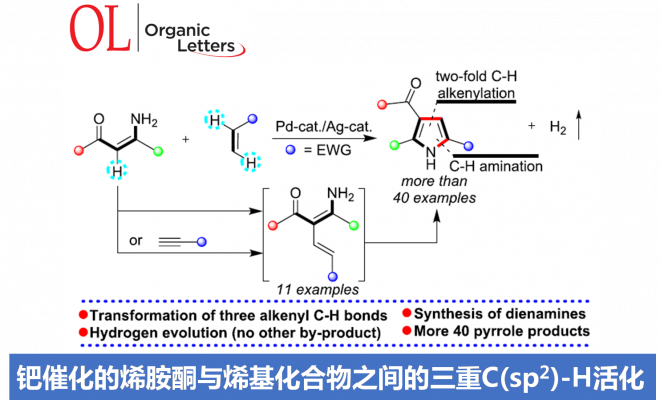
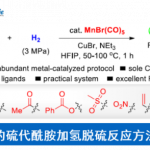
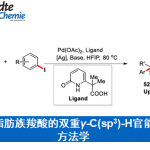
本文主要报道一种通过钯催化的烯胺酮 (enaminone)与烯基化合物之间的环化反应策略,进而成功完成一系列NH-游离吡咯 (NH-free pyrroles)分子的构建。上述策略中涉及三重C(sp2)-H键的活化。同时,通过氢气检测器 (hydrogen detector)能够进一步观察到上述的反应过程中,伴随氢气的析出。本文中,江西师范大学万结平与刘云云为共同通讯作者。
Pd-Catalyzed Triple-Fold C(sp2)−H Activation with Enaminones and Alkenes for Pyrrole Synthesis via Hydrogen Evolution
L. Fu, Y. Liu, J. Wan, Org. Lett. 2021, 23 , 4363. doi: 10.1021/acs.orglett.1c01301.

正文
芳香杂环骨架广泛存在各类天然产物以及药物分子中。其中,吡咯环的构建,在有机合成化学的发展中一直备受关注。例如较为经典的Paal-Knorr反应、Knorr反应以及Hantzsch反应方法学,对吡咯化学的发展曾作出巨大的贡献。然而,鉴于当代有机合成化学研究中对于设计分子多样性、可持续性以及安全性反应策略的高度需求,诸多课题组开始致力于发展全新的吡咯环构建的方法学策略[1]。其中,采用单重或多重C-H键活化方式进行的串联环化策略,已经发展成为构建吡咯骨架的一种全新策略。例如,Stuart等[2]报道将Rh(III)/Cu(II)催化剂促进的酰基烯胺与炔基化合物之间的环化反应方法学应用于吡咯骨架的构建。Glorius等[3]报道通过Pd(II)催化的亚胺分子内C-H烯基化策略,最终实现吡咯环骨架的构建。Ellmann等[4]报道Rh催化的烯酮衍生肟酯 (enone-derived oxime ester)与亚胺之间的烯基C-H官能团化方法学,进而实现一系列吡咯分子的合成。
在通过C-H活化或官能团化策略进行吡咯分子合成的研究中,烯胺底物的应用最为广泛。在适宜的过渡金属试剂催化下,较为稳定的烯胺,例如烯胺酮(enaminone)、烯胺酯 (enaminoester)以及烯胺腈 (enaminonitrile)等,目前已经应用于与炔基化合物、烯基化合物、重氮酯以及腙的相关偶联反应[5]或分子内C-H键官能团化[6]方法学策略的设计中,进而完成一系列多重官能团化吡咯分子的构建。然而,通过丙烯酸酯与稳定烯胺之间的偶联过程,构建相应的吡咯产物的策略,目前尚无相关的文献报道。
近年来,通过析氢 (hydrogen evolution)过程进行的C-H 以及杂原子-H键的脱氢转化策略 (dehydrogenative transformation),已经发展成为构建C-C或C-杂原子键的一种全新的合成设计方案[7]。尽管采用醇与氨基醇之间的脱氢过程,进行的吡咯化合物合成 (涉及氢气释放)已经取得较大进展[8]。然而,通过C-H键活化的方式进行相关的析氢过程,进而应用于吡咯环骨架构建的研究,迄今尚未有文献报道。本文中,作者受到烯基C-H键活化与官能团化的近期研究进展以及采用极性烯基化合物进行芳香环构建[9]的全新合成设计策略的启发,进而设计出一种采用简单烯胺酮与丙烯酸酯底物,通过三重 C(sp2)-H键与N-H键之间的析氢偶联 (hydrogen evolution-based coupling)过程,进而成功完成一系列吡咯衍生物的合成。
首先,作者采用烯胺酮1a与丙烯酸甲酯2a作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用10 mol% PdCl2作为催化剂,15 mol% AgOAc作为添加剂, 1 eq. CuBr2作为氧化剂,在DMF溶剂中,60 oC下反应,最终获得71%收率的吡咯产物3a。

在获得上述最佳反应条件后,作者开始对反应的底物适用范围进行考察(Scheme 1)。研究表明,一系列具有不同基团,例如甲基、乙基、正丁基、叔丁基以及苄基取代的丙烯酸酯底物,均能够顺利地参与上述成环反应,并以良好的收率获得相应的目标产物3a–3e以及3q–3u。之后,作者发现,一系列芳环中具有供电子与吸电子基团取代的芳基烯胺酮底物,均能够与上述的标准反应条件良好地兼容,并获得相应的吡咯产物3f–3z与3aa–3ac。同时,作者进一步观察到,芳环中具有多重取代基团的芳基烯胺酮底物,同样能够有效地完成上述的转化过程,并获得目标产物3al–3ao。此外,研究表明,稠合芳基以及杂芳基取代的烯胺酮底物,同样能够顺利地参与上述的偶联环化过程,进而完成一系列具有稠合芳基 (3ag与3ah)以及杂芳基 (3ai–3al)取代的吡咯分子的构建。
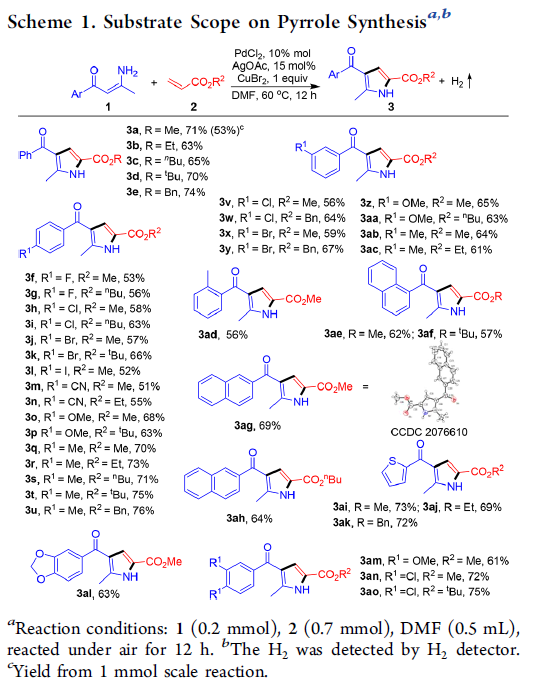
之后,作者发现,采用CuI作为催化剂时,烯胺酮与烷基丙炔酸酯底物4能够通过炔基C-H键的加成过程,选择性地获得一系列共轭双烯胺产物5 (Scheme 2)。同时,该小组发现,具有不同取代基团的烷基丙烯酸酯 (5a–5c)以及芳基不同位置中具有各类官能团取代的芳基烯胺酮底物 (5d–5g),均能够与上述标准催化反应条件有效地兼容。值得注意的是,呋喃基团取代的烯胺酮底物以及烯胺酯底物 (5h与5i)同样能够良好地与上述反应体系进行兼容。此外,上述反应策略同样能够应用于各类β-取代双烯胺产物 (5j与5k)的构建。

接下来,作者发现,双烯胺底物5在上述的标准反应条件下,能够顺利进行相应的分子内脱氢C-H胺化过程,最终获得一系列吡咯产物 3 (Scheme 3)。同时,在反应中过程中能够检测出氢气的产生。进而表明,双烯胺化合物5为烯胺-丙烯酸酯环化 (enamine-acrylate annulation)过程的关键中间体。
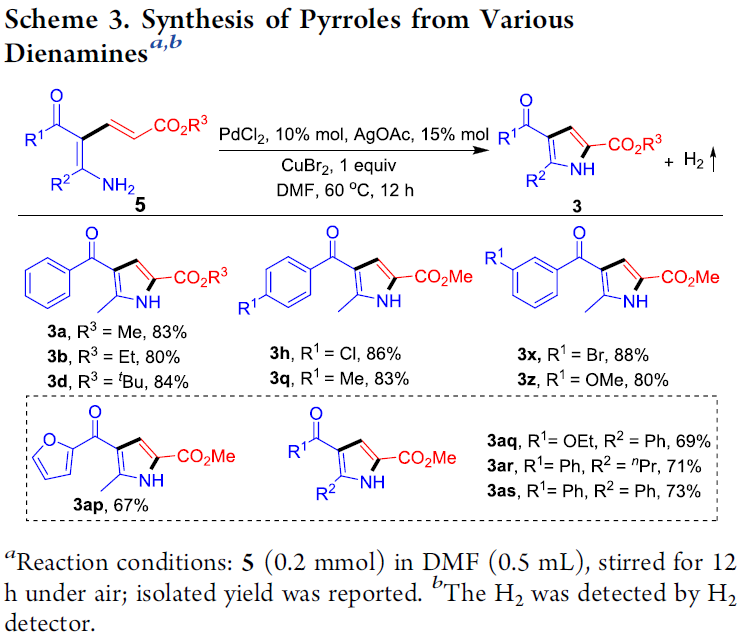
为进一步阐明合理的反应机理,作者进行一系列相关的控制实验研究。首先,作者发现,1a与2a在室温下进行反应时,能够获得吡咯3b与双烯胺5b (eq 1)。同时,作者进一步观察到,双烯胺底物5b在无金属试剂存在的条件下,并无法获得相应的吡咯产物3b,进而表明金属催化剂的存在,对于反应过程的顺利进行极为关键 (eq2)。

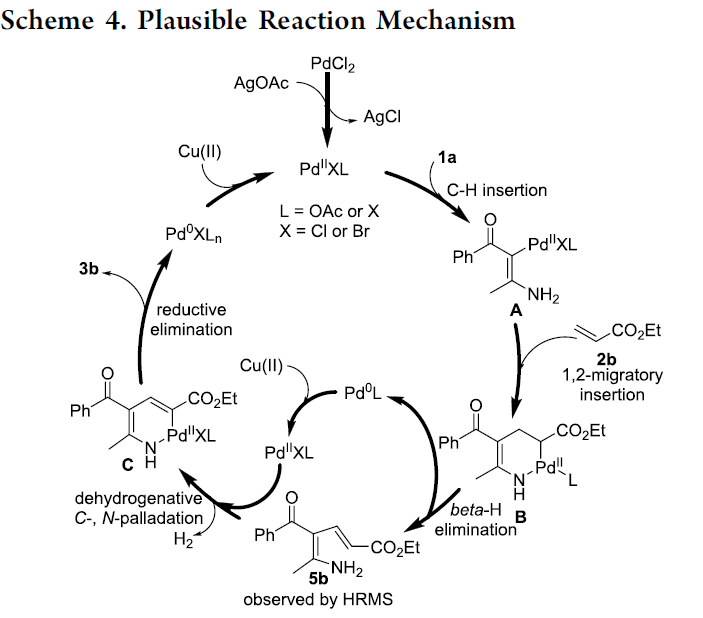
最后,作者提出一种可能的反应机理 (Scheme 4)。首先,通过AgOAc与PdCl2之间的离子交换过程,形成具有反应活性的PdIIXL中间体,PdIIXL与烯胺酮1a之间通过C-H键的插入过程,形成Pd(II)中间体A。之后,烯基化合物2b与中间体A进一步经历1,2-迁移插入过程,形成环钯配合物B,并通过B的β-H消除步骤,获得双烯胺中间体5b。接下来,通过双烯胺中间体5b与PdIIXL之间的脱氢C-,N-钯化过程 (dehydrogenative C-,N-palladation),形成环钯配合物C,最终,通过配合物C的还原消除过程,获得相应吡咯产物,并产生Pd0XLn。之后,通过Cu(II)氧化剂对Pd0XLn的氧化过程,使PdIIXL再生,进而完成相应的催化循环。
总结
本文主要报道一种通过钯催化剂促进的烯胺酮与烯基化合物之间的环化反应方法学,进而完成一系列吡咯衍生物的合成。并且,反应过程中涉及三重C(sp2)-H键的活化步骤。同时,上述策略具有底物应用范围广泛以及良好的官能团兼容性等优势。反应机理研究表明,双烯胺可能作为反应过程中的关键中间体。
参考文献
[1] (a) V. Estévez, M. Villacampa, J. C. Menéndez, Chem. Soc. Rev. 2014, 43, 4633. doi: 10.1039/C3CS60015G.(b) Y. Zhou, L. Zhou, L. T. Jesikiewicz, P. Liu, S. L. Buchwald, J. Am. Chem. Soc. 2020, 142, 9908. doi: 10.1021/jacs.0c03859.
(c) J. L i, B. Zhou, Y. Tian, C. Jia, X. Xue, F. Zhang, J. Ma, Org. Lett. 2020, 22, 9585. doi: 10.1021/acs.orglett.0c03621.
(d) B. Cheng, Y. Wang, T. Li, L. Lu, W. Xiao, J. Org. Chem. 2017, 82, 12134. doi: 10.1021/acs.joc.7b01931.
(e) Z. W. Gilbert, R. J. Hue, I. A. Tonks, Nat. Chem. 2016, 8, 63. doi: 10.1038/nchem.2386.
(f) Y. Liu, A. Parodi, S. Battaglioli, M. Monari, S. Protti, M. Bandini, Org. Lett. 2019, 21, 7782. doi: 10.1021/acs.orglett.9b02731.
[2] D. R. Stuart, P. Alsabeh, M. Kuhn, K. Fagnou, J. Am. Chem. Soc. 2010, 132, 18326. doi: 10.1021/ja1082624. [3] Z. Shi, M. Suri, F. Glorius, Angew. Chem. Int. Ed. 2013, 52, 4892. doi: 10.1002/anie.201300477. [4] Y. Lian, T. Huber, K. D. Hesp, R. G. Bergman, J. A. Ellmann, Angew. Chem. Int. Ed. 2013, 52, 629. doi: 10.1002/anie.201207995. [5] (a) S. Rakshit, F. W. Patureau, F. Glorius, J. Am. Chem. Soc. 2010, 132, 9585. doi: 10.1021/ja104305s.(b) J. Ke, C. He, H. Liu, M. Li, A. Lei, Chem. Commun. 2013, 49, 7549. doi: 10.1039/C3CC43682A.
(c) J. Chen, D. Chang, F. Xiao, G. Deng, J. Org. Chem. 2019, 84, 568. doi: 10.1021/acs.joc.8b02410.
(d) K. Luo, S. Mao, K. He, X. Yu, J. Pan, J. Lin, Z. Shao, Y. Jin, ACS Catal. 2020, 10, 3733. doi: 10.1021/acscatal.9b05360.
(e) M. Li, Y. Sun, Y. Xie, Y. Yu, F. Huang, H. Huang, Chem. Commun. 2020, 56, 11050. doi: 10.1039/D0CC04157B.
[6] (a) S. Yugandar, S. Konda, H. Ila, J. Org. Chem. 2016, 81, 2035. doi: 10.1021/acs.joc.5b02902.(b) X. Li, M. Chen, X. Xie, N. Sun, S. Li, Y. Liu, Org. Lett. 2015, 17, 2984. doi: 10.1021/acs.orglett.5b01281.
(c) N. E. Golantsov, A. S. Golubenkova, A. A. Festa, A. V. Varlamov, L. G. Voskressensky, Org. Lett. 2020, 22, 4726. doi: 10.1021/acs.orglett.0c01530.
[7] (a) X. Li, Z. Xin, S. Xia, X. Gao, C. Tung, L. Wu, Chem. Soc. Rev. 2020, 49, 9028. doi: 10.1039/D0CS00930J.(b) H. Wang, X. Gao, Z. Lv, T. Abdelilah, A. Lei, Chem. Rev. 2019, 119, 6769. doi: 10.1021/acs.chemrev.9b00045.
(c) Q. Liu, L. Wu, Nat. Sci. Rev. 2017, 4, 359. doi: 10.1093/nsr/nwx039.
[8] (a) P. Daw, S. Chakraborty, J. A. Garg, Y. Ben-David, D. Milstein, Angew. Chem. Int. Ed. 2016, 55, 14373. doi: 10.1002/anie.201607742.(b) D. Srimani, Y. Ben-David, D. Milstein, Angew. Chem. Int. Ed. 2013, 52, 4012. doi: 10.1002/anie.201300574.
(c) S. Qu, Y. Dang, C. Song, M. Wen, K. Huang, Z. Wang, J. Am. Chem. Soc. 2014, 136, 4974. doi: 10.1021/ja411568a.
[9] (a) Z. Gu, T. Zhu, J. Cao, X. Xu, S. Wang, S. Ji, ACS Catal. 2014, 4, 49. doi: 10.1021/cs400904t.(b) D. Hu, L. Yang, J. Wan, Green Chem. 2020, 22, 6773. doi: 10.1039/D0GC02806A.
(c) L. Yang, J. Wan, Green Chem. 2020, 22, 3074. doi: 10.1039/D0GC00738B.
(d) M. Zhao, Z. Ren, D. Yang, Z. Guan, Org. Lett. 2018, 20, 1287. doi: 10.1021/acs.orglett.7b04007.
(e) F. Zhang, Z. Qin, K. Kong, Y. Zhao, Y. Liu, Y. Li, Org. Lett. 2016, 18, 5150. doi: 10.1021/acs.orglett.6b02615.















No comments yet.