
作者:石油醚
引言
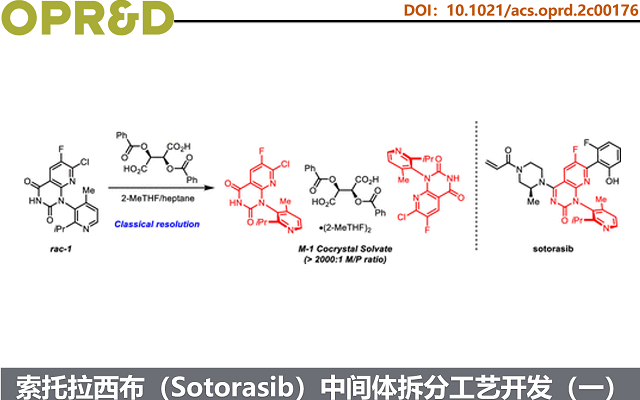
索托拉西布(Sotorasib)(Scheme 1)是一种靶向药物,它像一把设计精巧的”钥匙”,能够特异性地锁住并失活由KRAS G12C突变产生的异常蛋白,从而阻断癌细胞的生长信号。索托拉西布的药物活性成分含稠合双杂环核心和丙烯酰胺共价弹头,特异性靶向KRASG12C。分子含一个手性中心和一个阻转轴,其中(S,M)-二对映体生物活性最优。其中,轴手性极为稳定(旋转能垒42.3 kcal/mol,25°C下半衰期>100亿年)。
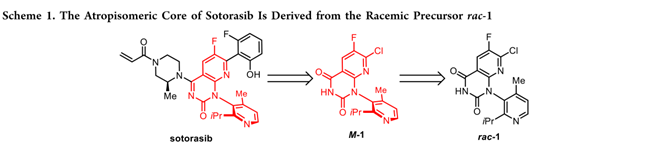
药物研发初期,索托拉西布通过多步合成制备,且关键中间体为rac–1。在临床前及早期临床阶段,采用手性色谱(包括标准柱色谱和模拟移动床)成功拆分 rac–1,获得目标构型中间体 M–1,支撑毒理学评价和首次人体试验。但该方法周期长、成本高、溶剂消耗大,导致工艺质量强度高、供应链复杂,不适用于商业化生产。因此,转向开发高效手性拆分工艺,以实现 M–1 的稳定、无需色谱分离商业供应,保障索托拉西布的长期量产。
正文
中间体rac-1的合成路线
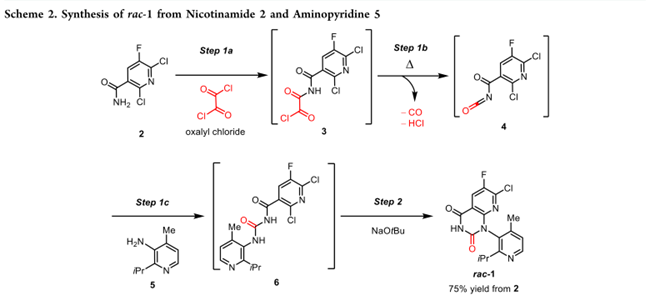
rac–1 通过烟酰胺 2 的多步串联反应制备(scheme 2)。即2在DCM中与草酰氯反应生成中间体 3;加热促使 3 同步脱羧、脱氯化氢,得到酰基异氰酸酯 4。4 不经分离,直接与氨基吡啶 5 反应生成酰基脲 6。随后经水相处理、溶剂置换为 2-甲基四氢呋喃(2-MeTHF),再加入叔丁醇钠(NaOtBu)引发分子内环合,析出结晶得 rac–1。该工艺已成功放大至 2 投料量 >100 kg,总收率 75%。
中间体M-1的手性拆分工艺开发
首先,作者采用高通量实验筛选经典拆分条件(Scheme 3),评估其对映体分离可行性。首轮筛选涵盖手性拆分试剂/溶剂组合(>275条件),均未获得对映体富集的 1。另筛选 100 种酸/溶剂组合,聚焦初始实验未覆盖的替代溶剂。所得固体均经粉末X射线衍射(PXRD)鉴定晶型。结果表明:多数条件下析出 rac–1 的不同晶型;部分体系生成 rac–1 与拆分剂的混合物。进而证实大多数拆分剂无法与底物形成有效手性识别复合物,因而无法实现对映体拆分。
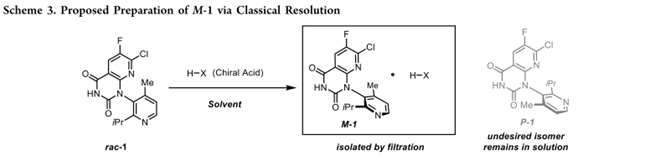
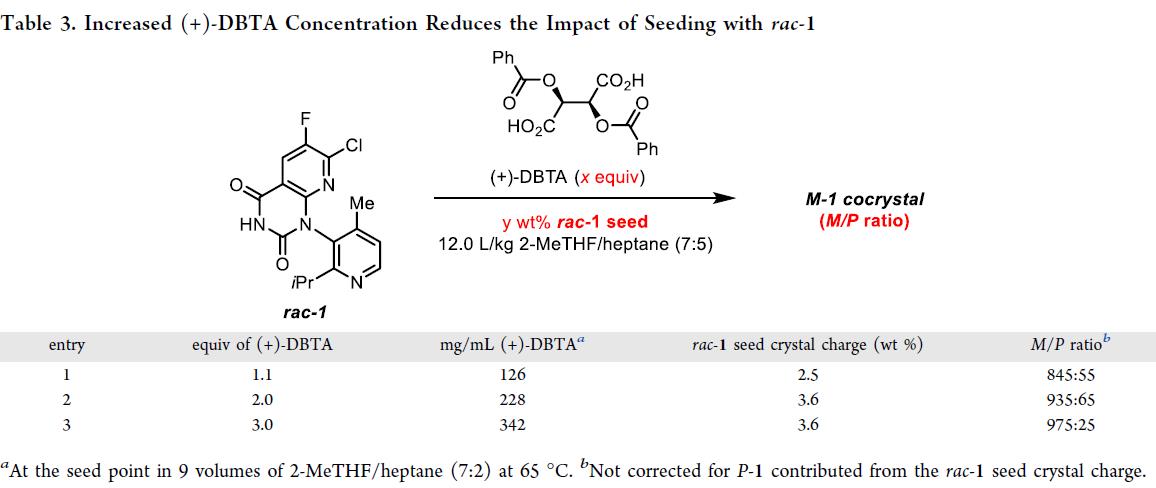
进一步通过实验并结合PXRD,表明三种酸在不同溶剂中生成了新物种:(1S)-(−)-樟脑酸、D-(+)-苹果酸和(+)-2,3-二苯甲酰-D-酒石酸[(+)-DBTA]。手性 HPLC 进一步证实:仅(+)-DBTA 实现了有效拆分,得到 M/P = 81:19(Table 1)。后续筛选 20 种替代溶剂,均未提升拆分效率。因此转向优化 2-MeTHF/庚烷比例,以提高 M/P 比值并最大化 M–1/(+)-DBTA 复合物收率。

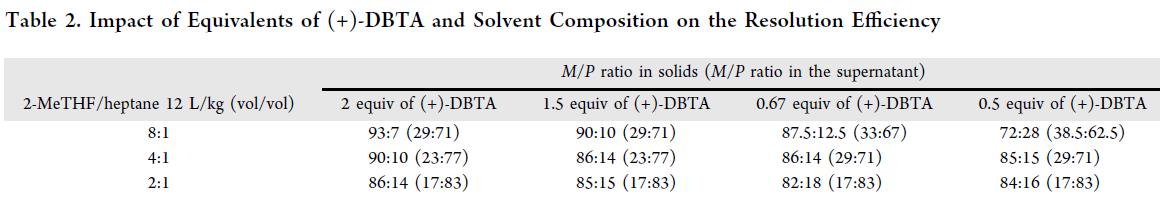
同时,考察(+)-DBTA 用量对选择性的影响(Table 2)。实验表明:M/P 比值随(+)-DBTA 当量增加而升高,但随庚烷比例升高而降低。因此,在高比例 2-MeTHF 下提高(+)-DBTA 用量可最大化 M/P,但会牺牲回收率。
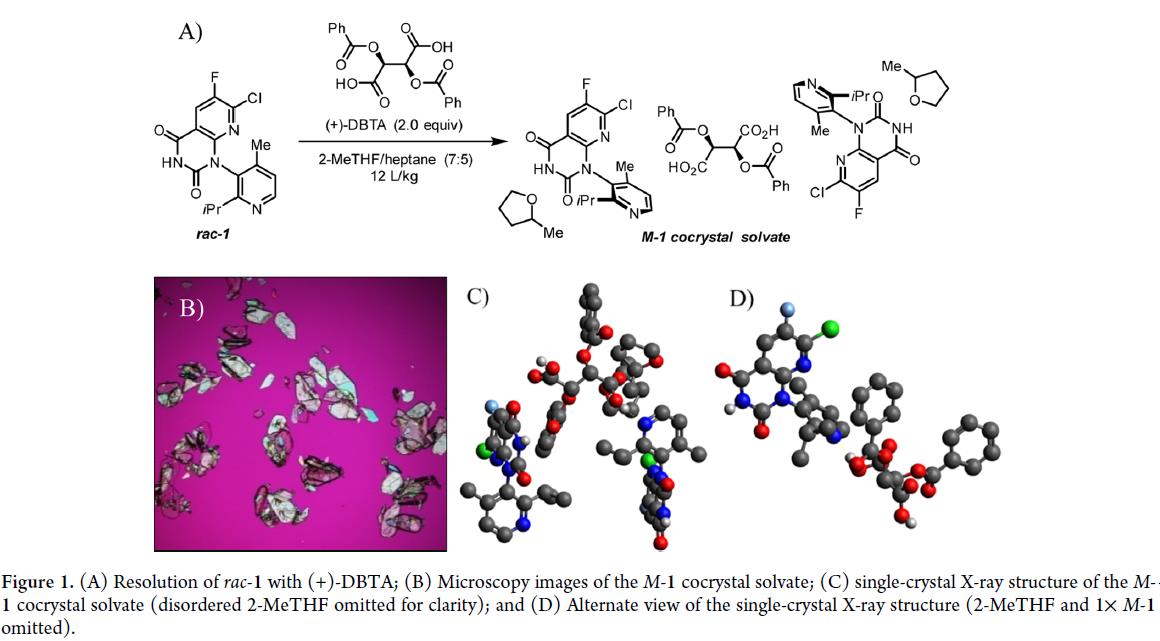
此前所有实验均采用“浆液到浆液”模式——即 rac–1、(+)-DBTA 与 2-MeTHF/庚烷一次性投料。为提升效率,改用受控的“溶液到浆液”工艺:先在 75 °C 下用纯 2-MeTHF(7.0 L/kg)完全溶解 rac–1 和(+)-DBTA(2 equiv),再于 65 °C 加入庚烷(2.0 L/kg)反溶剂及晶种析出浆液;随后冷却至 20 °C 并补加庚烷(3 L/kg)深度脱饱和,最终获得 M/P >99.9:0.1 的产物,分离收率 44%(理论上限 50%)。该结果证实,精准控制溶解、成核与结晶过程可实现高效拆分。NMR 显示产物组成为 M-1: (2-MeTHF):(+)-DBTA(2:2:1),单晶 X 射线结构证实其为三组分氢键共晶溶剂化物(Figure 1)。产物中 2-MeTHF 含量受干燥条件影响,通常为 2–3 equiv。
进一步为明确低 M/P 样品的组成,开展 1H NMR 分析,结果显示其 DBTA 含量低于理论值(1.0 equiv per 1)。尝试将纯化后的 P–1 置于标准拆分条件下,未能获得 P–1/(+)-DBTA 共晶溶剂化物,进而表明低 M/P 源于 rac–1 与 M–1/(+)-DBTA 复合物的共结晶。鉴于完全拆分所需 (+)-DBTA 的理论下限为 0.5 equiv(相对于 rac–1),提高 (+)-DBTA 浓度有望加速 M-1/(+)-DBTA 在溶液中的形成,并优先驱动其结晶。
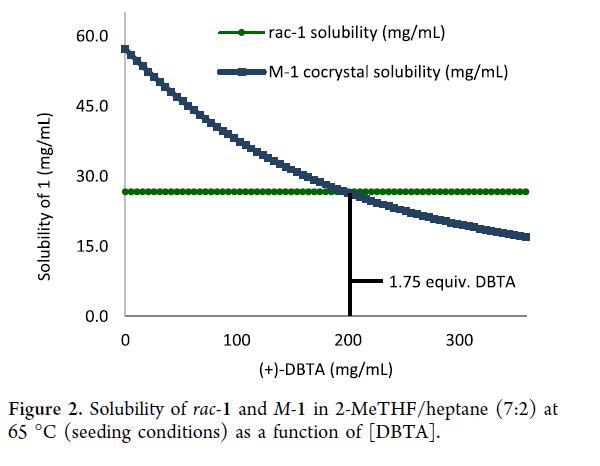
接下来,优化(+)-DBTA 用量以提升工艺稳健性,并利用各组分溶解度数据构建热力学模型。建模条件:2-MeTHF/庚烷 = 7:2(9.0 L/kg),晶种温度 65 °C。模型预测:rac–1 溶解度不受(+)-DBTA 浓度影响(Figure 2),而 M–1 共晶溶剂化物的相对溶解度随(+)-DBTA 浓度升高而显著降低,即当浓度 >200 mg/mL(≈1.75 equiv)时,其结晶在热力学上占优;同时,更高浓度可加速 M–1 复合,抑制 rac–1 共结晶。该模型经三组不同(+)-DBTA 浓度的实验验证:以 rac–1 为晶种模拟最差工况,结果与模型一致——(+)-DBTA 浓度越高,M/P 比值越高(Table 3)。据此,选定 3.0 equiv(+)-DBTA 作为工艺标准用量,确保高 M/P 产物稳定输出。
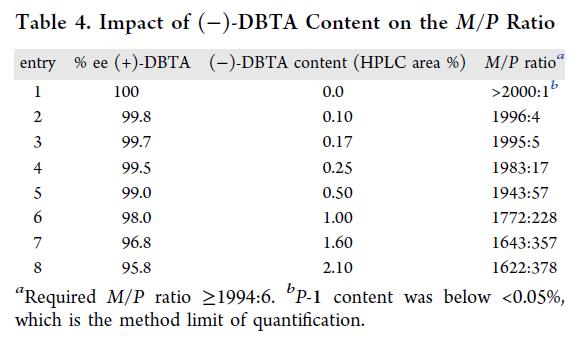
随后,聚焦(+)-DBTA 的对映体纯度要求。鉴于用量远超理论最小值(3.0 vs 0.5 equiv),痕量(−)-DBTA 可能显著劣化产物 ee。加标实验表明:仅含 0.10%(HPLC 面积比)(−)-DBTA 即降低选择性;更高含量则严重恶化 M/P 比(Table 4)。因此,将(+)-DBTA 的杂质限值设定为 ≤0.10%(LC 面积比),即 ≥99.8% ee,以保障产物 M/P ≥1994:6(≥99.4% ee)。
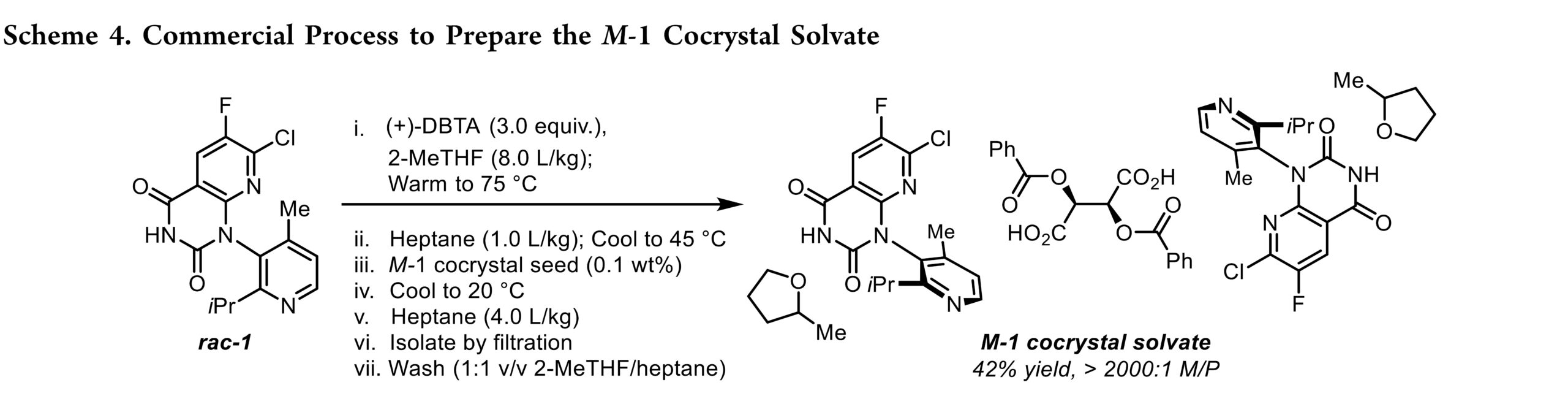
基于上述分析,实施三项关键优化:(1)2-MeTHF 用量由 7.0 L/Kg增至 8.0 L/Kg,显著改善浆料流动性并消除结垢;(2)种子点庚烷含量由 2.0 L/Kg降至 1.0 L/Kg,确保热循环下可完全再溶解;(3)相应将晶种加入温度由原值下调至 45 °C,防止晶种溶解。最终商业化工艺条件见scheme 4。即rac–1(1.0 equiv)与(+)-DBTA(3.0 equiv)溶于 2-MeTHF(8.0 L/kg),75 °C 加热至全溶。随后,加入庚烷(1.0 L/kg),降温至 45 °C,投入 M-1 共晶溶剂化物晶种(0.1 wt%),陈化 30 min;4 h 内梯度冷却至 20 °C,再陈化 30 min;1 h 内补加庚烷(4.0 L/kg);过滤,滤饼用 2-MeTHF/庚烷(1:1 v/v,2 × 3.0 L/kg)淋洗;25 °C 真空干燥,得白色结晶性 M–1 共晶溶剂化物(42% 收率,M/P >2000:1)。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/


















No comments yet.