
本文作者:Summer
导读
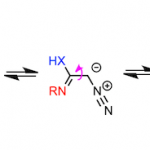
新加坡南洋理工大学池永贵教授课题组与贵州中医药大学田维毅课题组共同报道首例通过NHC催化的(苯并)咪唑衍生醛亚胺 ((benz)imidazole-derived aldimine)与靛红 (isatin)衍生物或三氟苯乙酮 (trifluoroacetophenone)之间的不对称 [4+2]环加成反应方法学,以良好至优秀的收率与对映选择性获得一系列杂环产物 3。该方法学是将氧化型氮杂-Breslow中间体 (oxidized aza-Breslow intermediate)应用于不对称合成研究的首次尝试。同时,作者假设反应过程通过一种新型的活性中间体,即三氮杂二烯中间体 (triaza diene intermediate) III进行。此外,反应机理研究与DFT计算表明,在一系列反应机理步骤中,[4+2]环化步骤为协同异步加成 (concerted asynchronous addition)过程,并且,该步骤为反应过程的对映选择性决定步骤。
Carbene-Catalyzed Activation of Remote Nitrogen Atoms of (Benz)imidazole-Derived Aldimines for Enantioselective Synthesis of Heterocycles
X. Yang, Y. Xie, J. Xu, S. Ren, B. Mondal, L. Zhou, W. Tian, X. Zhang, L. Hao, Z. Jin, Y. R. Chi, Angew. Chem. Int. Ed. 2021, 60, 7906. doi: 10.1002/anie.202016506.
正文
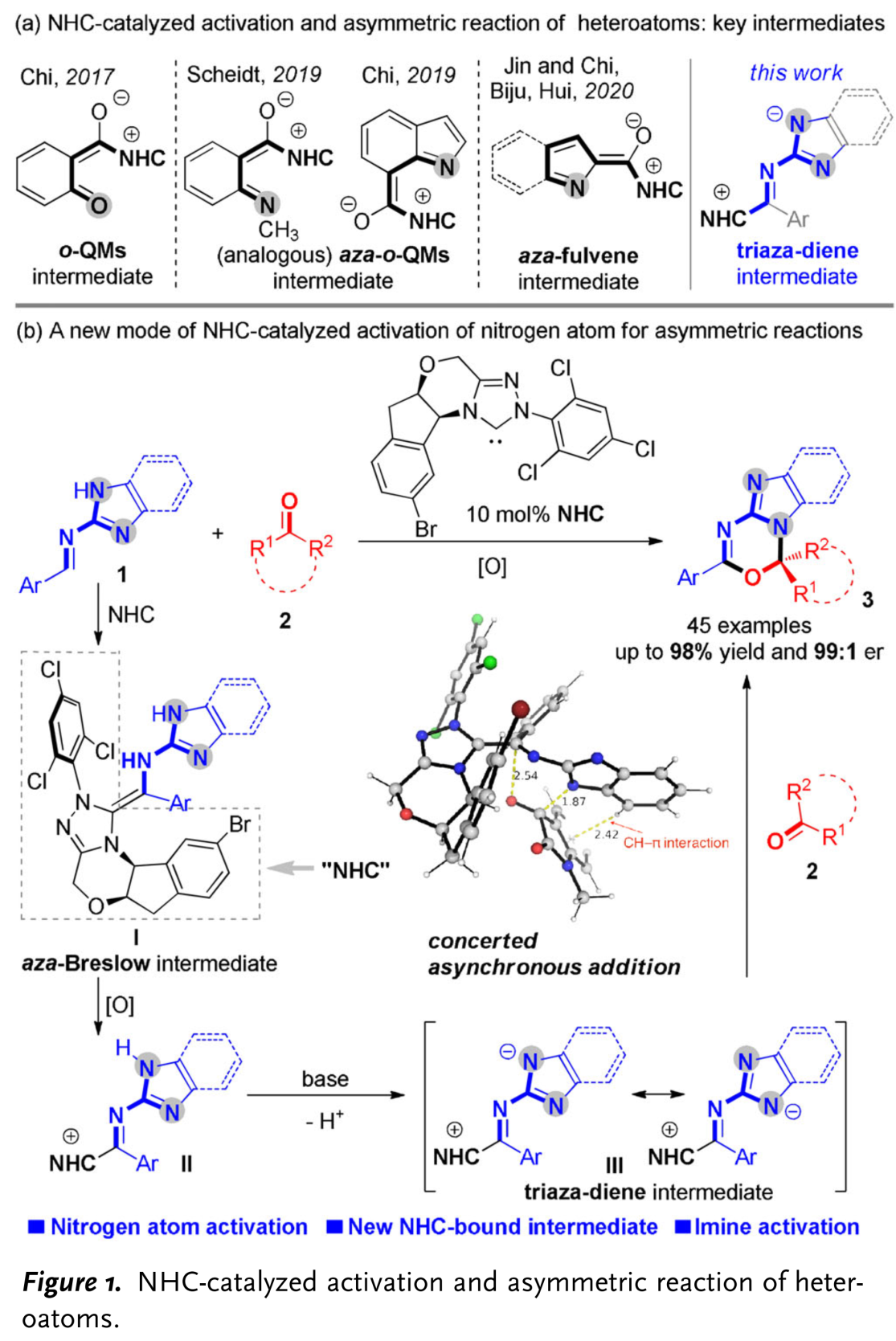
含有sp3杂原子的有机分子作为亲核试剂,通常能够与NHC有机催化剂形成的亲电性 (不饱和)酰基唑中间体 ((unsaturated) acyl azolium intermediates)进行有效的反应[1]。然而,通过NHC有机催化剂对杂原子亲核试剂进行催化活化的不对称合成方法学研究较少有文献报道。目前,相关报道主要涉及:采用azolium salt (NHC 前催化剂,NHC pre-catalysts)作为非共价催化剂 (non-covalent catalyst)促进的由胺或硫醇亲核试剂参与的对映选择性合成转化[2]、NHC催化的通过磺酰亚胺S-N键断裂产生的亚磺酸负离子 (sulfinic anion)进行的对映选择性磺化反应[3]、通过NHC催化剂与水杨醛作用,形成的o-QM (quinone methide)中间体而进行的不对称环化反应[4]、采用NHC催化剂与N-甲基靛红酸酐 (N-methylisatoic anhydride)作用产生的aza-o-QM中间体而进行的对映选择性N-亲核加成反应 (enantioselective N-nucleophilic addition)反应 (Figure 1a) [5]以及通过NHC催化剂与吲哚甲醛作用,形成的氮杂富烯 (aza-fulvene)中间体而进行的不对称N-H键官能团化 (asymmetric N-H functionalization)[6]。在上述采用NHC催化剂参与的杂原子不对称官能团化方法学 (Figure 1a)的研究报道中,反应活性的亲核性杂原子位于底物中羰基官能团的γ-位置。因此,这类中间体均可以视为将γ-碳原子采用杂原子进行取代的乙烯基烯醇负离子中间体的类似物。基于上述研究报道,新加坡南洋理工大学的池永贵教授课题组与贵州中医药大学田维毅课题组通过选择NHC有机催化剂,共同开发出一种在不对称合成反应中,对杂原子进行活化的新策略 (Figure 1b)。其反应机理主要涉及:首先,NHC催化剂与(苯并)咪唑衍生醛亚胺结合,形成氮杂-Breslow中间体 (aza-Breslow intermediate) I。之后,在氧化剂作用下,将中间体 I转化为缺电子的中间体 II。接下来,在碱存在下,II 通过质子转移过程,转化为三氮杂二烯 (triaza diene)中间体 III。最终,III与靛红衍生物2进行不对称[4+2]环加成反应,并以良好至优秀的收与对映选择性获得一系列手性杂环产物 3。同时,通过DFT计算表明,上述的[4+2]环加成步骤为协同异步加成过程 (concerted asynchronous addition process)。同时,作者提出一种新型的NHC催化中间体,即三氮杂二烯中间体 III。这一方法学的发展,是将氧化型氮杂-Breslow中间体 (oxidized aza-Breslow intermediate)应用于不对称合成方法学的首次尝试。
(图片来源:Angew. Chem. Int. Ed.)
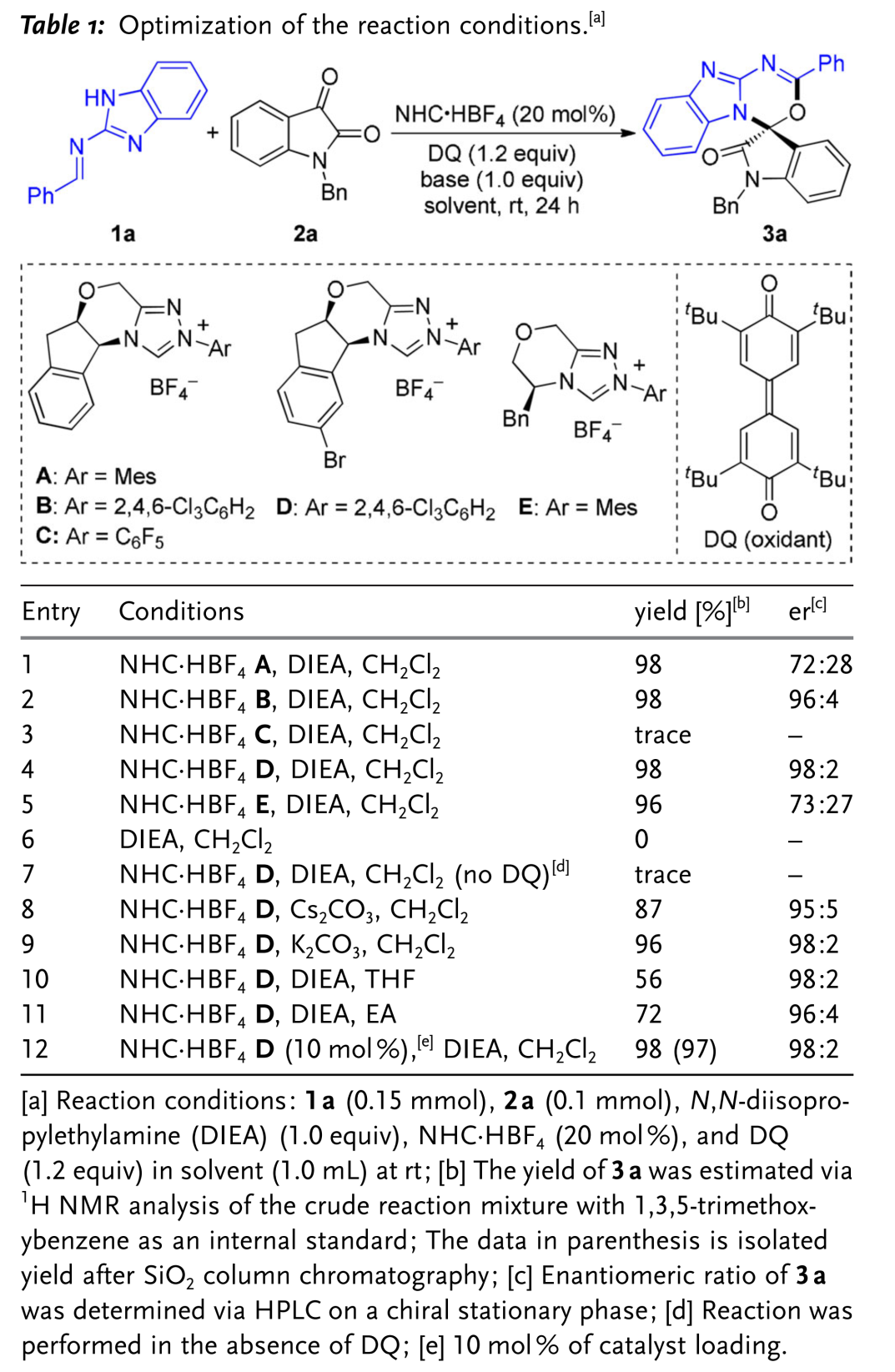
首先,作者采用 (苯并)咪唑衍生的醛亚胺1a与靛红衍生物2a作为模板底物,通过对催化剂、溶剂以及碱等条件进行筛选,确定最佳的反应条件 (Table 1)为:采用10mol % NHC·HBF4 D作为催化剂,1.0 eq. DIEA作为碱,1.2 eq. DQ作为氧化剂,CH2Cl2作为反应溶剂,在室温条件下,反应时间为24 h。能够以97 %的收率与96 %的对映选择性获得相应产物3a。
(图片来源:Angew. Chem. Int. Ed.)
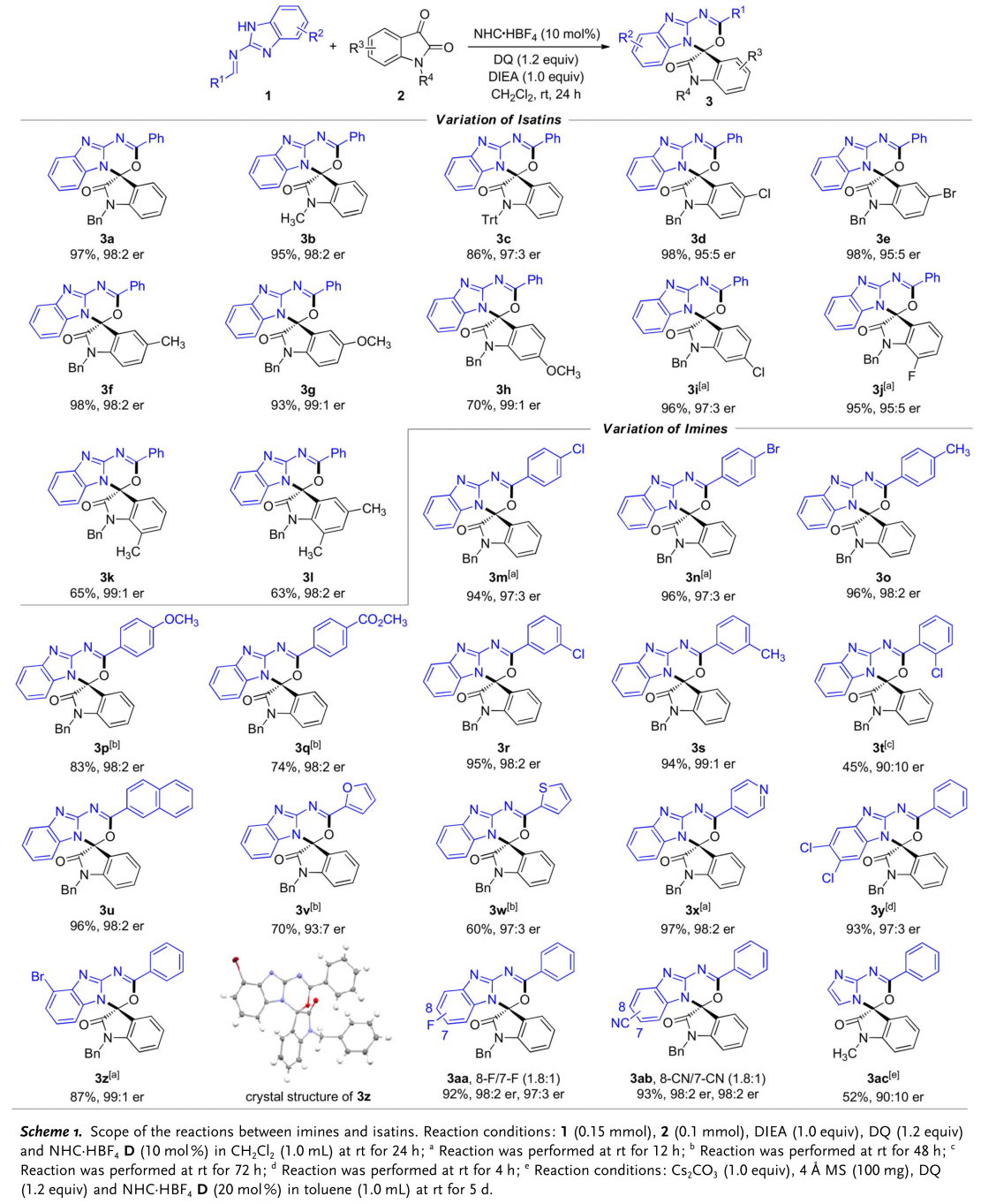
之后,在最上述的最佳反应条件下,作者对于各类 (苯并)咪唑衍生醛亚胺与靛红衍生物的底物适用范围进行考察 (Scheme 1)。研究表明,一系列N-取代以及芳基取代的靛红衍生物,均能够较好地与上述反应条件兼容,并能够以良好至优秀的收率与优秀的对映选择性获得相应手性产物。并且,作者发现,各类对位、间位苯基取代、2-萘基取代与杂芳基取代的醛亚胺以及通过5,6-二氯苯并咪唑基团与4-溴苯并咪唑基团保护的醛亚胺均能够与N-苄基取代的靛红进行有效的反应,并以良好至优秀的收率与优秀的对映选择性获得相应手性产物。其中,3z的绝对构型通过X-射线晶体学分析确定。而对于其它5-取代苯并咪唑保护的醛亚胺,尽管能够获得较高的反应收率与对映选择性,然而,却获得7-与8-取代的两种区域异构体的混合物 (3aa 与3ab,区域异构体的比例为1.8:1),即无法获得优良的区域选择性。此外,作者进一步发现,采用咪唑保护的醛亚胺底物,仅能够以中等的收率与良好的对映选择性获得相应手性产物。
(图片来源:Angew. Chem. Int. Ed.)
接下来,为进一步阐明上述方法学的普适性与合成应用价值,作者进一步对醛亚胺与一系列非环酮底物之间的反应进行考察。首先,作者选择醛亚胺1与三氟苯乙酮4作为模型底物 (Scheme 2),确定其最佳的反应条件为:采用20 mol % NHC·HCl F作为催化剂,1.0 eq. Cs2CO3作为碱,100 mg 4Å分子筛作为添加剂,1.2 eq. DQ作为氧化剂,t–BuOH作为反应溶剂,在室温条件下,反应时间为48 h,能够以92%的收率与88% 的对映选择性获得相应产物5a。接下来,作者进一步观察到,一系列三氟芳乙酮、三氟甲基脂肪酮以及α-酮酸酯底物均能够有效地与醛亚胺1进行反应,并以良好至优秀的收率与对映选择性获得一系列手性三氟甲基化的杂环产物。其中,5n的绝对构型通过X-射线晶体学分析确定。

(图片来源:Angew. Chem. Int. Ed.)
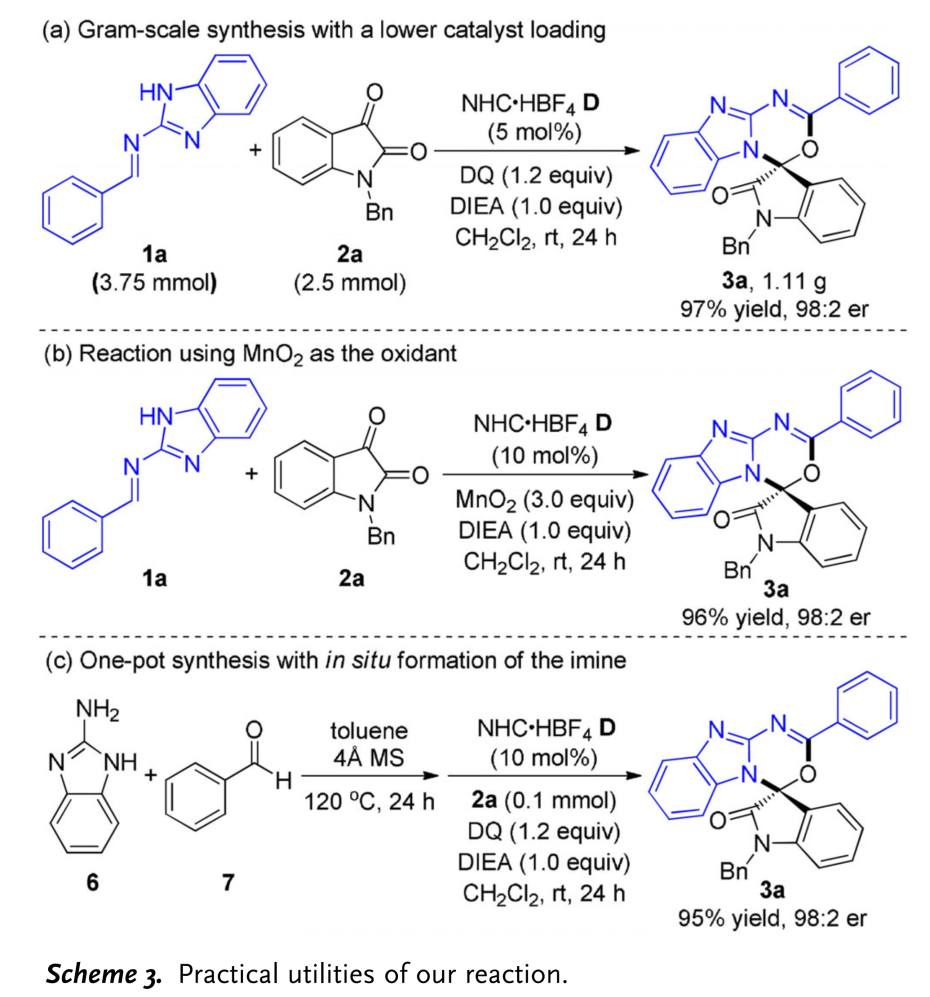
同时,该小组进一步研究表明,将2a的用量扩大至2.5mmol时,上述的[4+2]环加成反应同样能够有效地进行,并获得优良的反应收率与对映选择性 (Scheme 3a)。此外,研究发现,有机氧化剂DQ能够通过更为廉价的无机氧化剂MnO2进行替代 (Scheme 3b)。同时,该小组将胺6与苯甲醛7原位产生的亚胺1进一步采用一锅反应,最终获得相应目标产物3a (Scheme 3c)。
(图片来源:Angew. Chem. Int. Ed.)
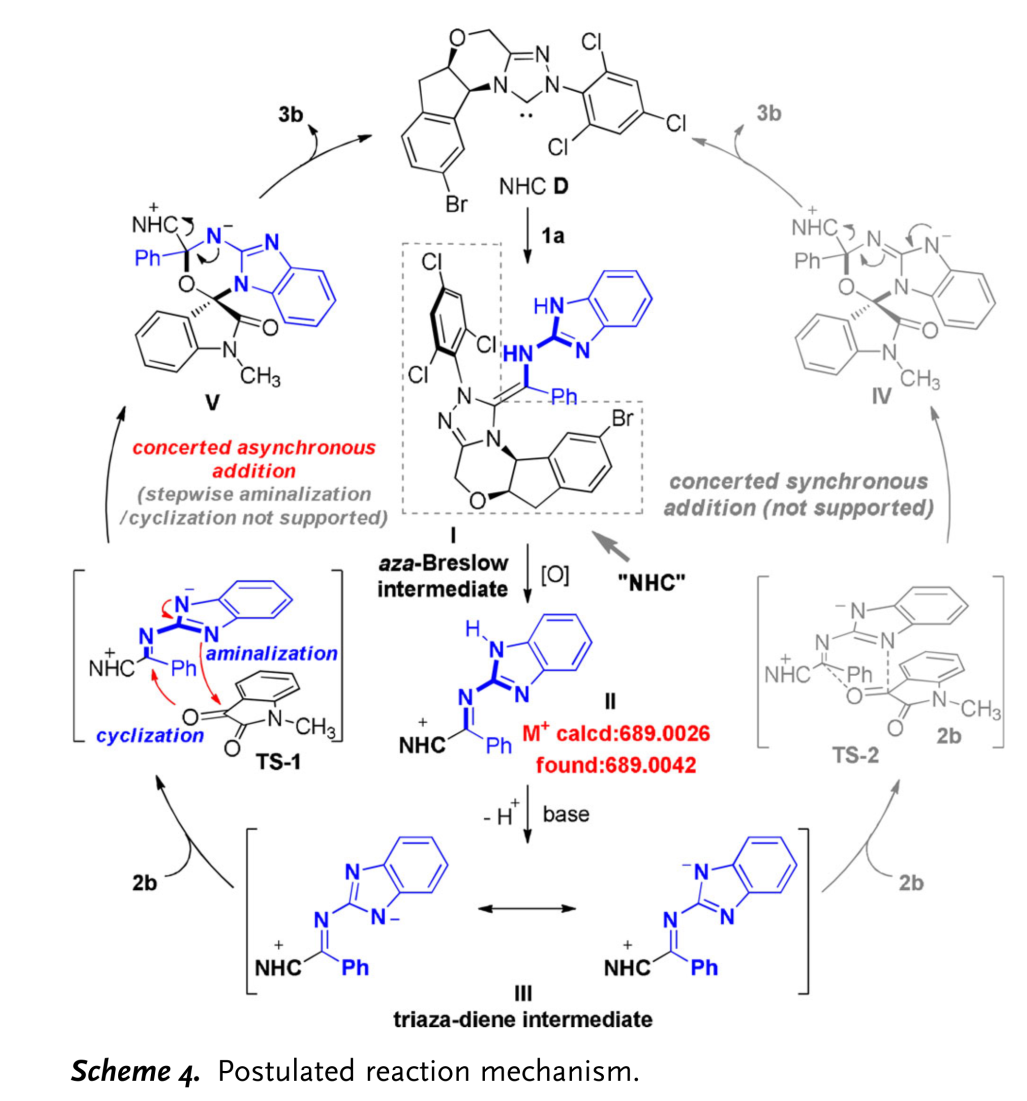
结合实验观察[7]与DFT计算,作者提出一种合理的反应机理 (Scheme 4):首先,NHC催化剂D与2-氨基 (苯并)咪唑衍生的亚胺1a结合,形成氮杂-Breslow中间体I。之后,在氧化剂DQ作用下,中间体I转化为缺电子的中间体II (中间体II能够通过HRMS检测出,详见supporting information)。接下来,在碱作用下,II通过去质子化过程,进一步转化为三氮杂二烯中间体III。最后,III与靛红衍生物2b通过TS-1过渡态,进而通过不对称[4+2]环加成过程,形成手性产物3b,同时,使NHC催化剂D再生。其中, DFT计算表明,反应过程中的[4+2] 环加成步骤为协同异步加成过程。
(图片来源:Angew. Chem. Int. Ed.)
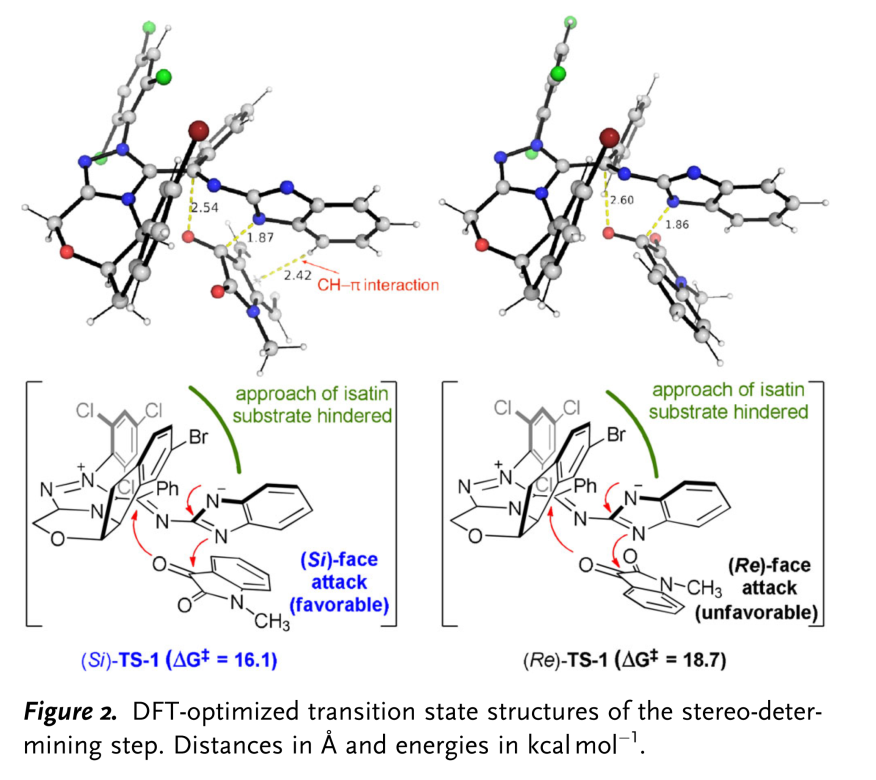
此外,作者通过DFT计算,并结合对立体选择性决定过渡态 (stereo-determining transition state)中FMO与NCI (non-covalent interaction)以及DI-AS (distortion-interaction /activation strain) [8]的研究,进一步证实,反应过程中控制对映选择性的关键因素为[4+2]环化步骤 (Figure 2 and supporting information)。
(图片来源:Angew. Chem. Int. Ed.)
小结
新加坡南洋理工大学池永贵教授课题组与贵州中医药大学田维毅课题组共同报道了首例通过NHC催化的 (苯并)咪唑衍生物与靛红衍生物或三氟苯乙酮之间的不对称[4+2]环加成反应方法学。该方法学能够以良好至优秀的收率与对映选择性获得一系列手性杂环产物3。同时, DFT计算表明,在一系列反应机理步骤中,[4+2]环化步骤为协同异步加成过程,并且为反应过程中的对映选择性决定步骤。
参考文献
- a) S. Mondal, S. R. Yetra, S. Mukherjee, A. T. Biju, Chem. Res. 2019, 52, 425. doi: 10.1021/acs.accounts.8b00550.b) C. Zhang, J. F. Hooper, D. W. Lupton, ACS Catal.2017,7, 2583. doi: 1021/acscatal.6b03663.c) X. Wu, B. Liu, Y. Zhang, M. Jeret, H. Wang, P. Zheng, S. Yang, Song, Y. R. Chi, Angew. Chem. Int. Ed. 2016, 55, 12280. doi: 10.1002/anie.201606571.d) A. Ghosh, S.Barik, A. T. Biju, Lett. 2019, 21, 8598.doi: 10.1021/acs.orglett.9b03188.e) S. Wu, C. Liu, G. Luo, Z. Jin, P. Zheng, Y. R. Chi, Chem. Int. Ed.2019, 58, 18410.doi: 10.1002/anie.201909479.f) D. Meng, Y. Xie, Q. Peng, J. Wang, Lett.2020, 22, 7635.doi: 10.1021/acs.orglett.0c02832.
- a) Wang, J. Chen, Y. Huang, Angew. Chem. Int. Ed. 2015, 54, 15414. doi: 10.1002/anie.201508371.b) J. Chen, S. Meng, L. Wang, H. Tang, Y. Huang, Sci.2015, 6, 4184. doi: 10.1039/C5SC00878F.
- Jin, J. Xu, S. Yang, B. Song, Y. R. Chi, Angew. Chem. Int.Ed. 2013, 52, 12354. doi: 10.1002/anie.201305023.
- Chen, H. Wang, K. Doitomi, C. Y. Ooi, P. Zheng, W. Liu,H. Guo, S. Yang, B. Song, H. Hirao, Y. R. Chi, Nat. Commun. 2016, 8, 15598. doi: 10.1038/ncomms15598.
- Lee, J. L. Zhu, T. Feoktistova, A. C. Brueckner, P. Cheong,K. A. Scheidt, Angew. Chem. Int. Ed. 2019, 58, 5941. doi: 10.1002/anie.201900600.
- a) Liu, G. Luo, X. Yang, S. Jiang, W. Xue, Y. R.Chi, Z. Jin, Angew. Chem. Int. Ed. 2020, 59, 442. doi: 10.1002/anie.201912160.b) X. Yang, G. Luo, L. Zhou, B. Liu, X. Zhang, H. Gao, Z. Jin, Y. R. Chi, ACS Catal.2019, 9, 10971. doi: 1021/acscatal.9b03163.c) Balanna, K. Madica, S. Mukherjee, A. Ghosh, T. Poisson, T. Besset, G. Jindal, A. T. Biju, Chem.–Eur. J.2020, 26, 818. doi: 10.1002/chem.201905177.d) Wang, Z. Li, J. Zhang, X. Hui, Org. Chem. Front.2020, 7, 1647. doi: 10.1039/D0QO00237B.
- Wang, Z. Fu, W. Huang, Org. Lett.2017, 19, 3362. doi: 10.1021/acs.orglett.7b01195.
- a) D. H. Ess, K. N. Houk, Am. Chem. Soc.2007, 129, 10646. doi: 10.1021/ja0734086.b) F. M. Bickelhaupt, K. N. Houk, Chem. Int. Ed. 2017, 56, 10070.doi:10.1002/anie.201701486.c) F. M. Bickelhaupt, Comput. Chem. 1999, 20, 114. doi:10.1002/(SICI)1096-987X(19990115)20:1<114::AID-JCC12>3.0.CO;2-L.d) I. Fernández, F. M. Bickelhaupt, Soc. Rev. 2014, 43, 4953. doi:10.1039/C4CS00055B.e) L. P. Wolters, F. M. Bickelhaupt, Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2015, 5, 324. doi:1002/wcms.1221.f) P. Vermeeren, S. C. C. van der Lubbe, C. F. Guerra, F. M. Bickelhaupt, T. A. Hamlin, Protoc. 2020, 15, 649. doi: 10.1038/s41596-019-0265-0.


















No comments yet.