
本文作者:杉杉
导读
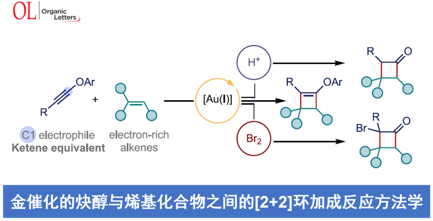
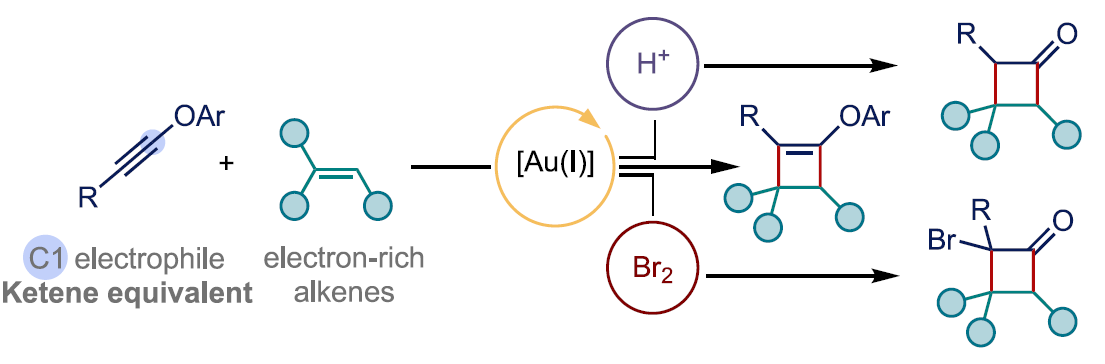
近日,西班牙ICIQ (Institut Català d’Investigació Química)的A. M. Echavarren课题组在Org. Lett.中发表论文,报道一种通过金(I)催化剂促进的炔醇醚 (ynol ethers)与烯基化合物之间的[2+2]环加成反应方法学,进而成功实现一系列环丁酮衍生物的构建。同时,该小组发现,内芳基炔醇醚与富电子的苯乙烯分子之间,则优先进行相应的[4+2]环加成过程,进而顺利实现各类色烷分子的构建。
Synthesis of Cyclobutanones by Gold(I)-Catalyzed [2+2] Cycloaddition of Ynol Ethers with Alkenes
M. Zanini, A. Cataffo, A. M. Echavarren, Org. Lett. 2021, ASAP doi: 10.1021/acs.orglett.1c03499.
正文
作为环羰基化合物中能够稳定存在的最小骨架单元,环丁酮类化合物已经成为有机合成方法学研究中极为关键的砌块。因此,构建环丁酮分子的相关策略研究,一直以来备受合成化学家的广泛关注。目前,相关的反应策略主要涉及:烯酮与烯基化合物之间的[2+2]环加成[1]-[2] (Scheme 1A)、取代环丙烷或环丙醇的扩环反应[3]、形式[3+1]环加成反应[4]、金-卡宾前体的C-H插入反应[5]、Co(II)催化的自由基1,4-C-H烷基化反应[6]、环丁酮衍生物的官能团化反应[7]以及金或铂催化的烯-炔酰胺环异构化反应方法学[8]-[9]。
这里,作者受到Kozmin课题组对于银催化的硅基炔醇醚与α,β-不饱和酮之间的[2+2]环加成反应方法学[10] (Scheme 1B)以及本课题组前期对于金催化的炔基与烯基化合物之间的[2+2]环加成反应方法学[11]-[14]相关研究的启发,本文中,作者成功设计出一种通过金催化剂促进的芳基炔醇醚与烯基化合物之间的[2+2]环加成反应方法学,进而有效地实现一系列环丁酮分子的构建 (Scheme 1B)。同时,研究发现,内芳基炔醇醚与烯基化合物之间,则优先进行相应的[4+2]环加成过程,进而获得一系列重要的色烷分子。

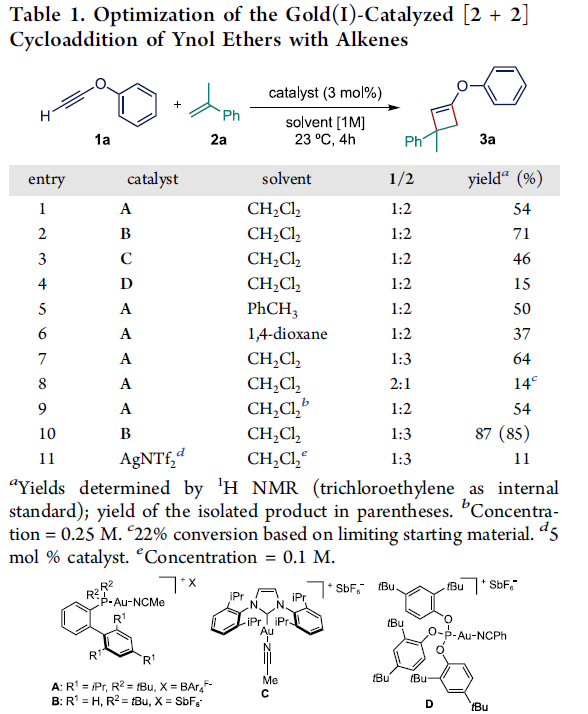
首先,作者采用苯基炔醇醚1a与α-甲基苯乙烯2a作为模型底物,进行相关环加成反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用金配合物B作为催化剂,二氯甲烷作为反应溶剂,反应温度为23 oC,最终获得87%收率的环加成产物3a。
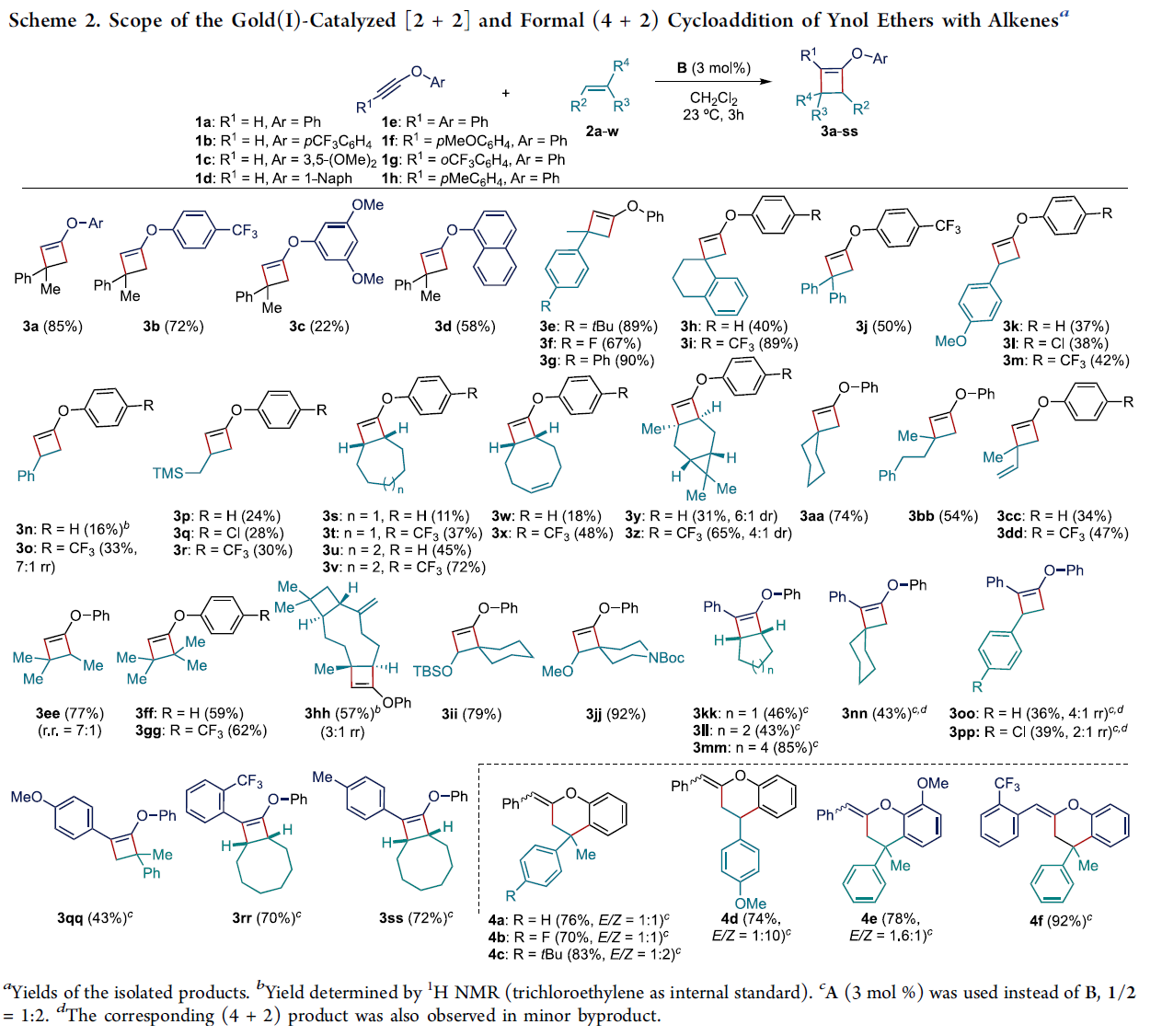
在上述的最佳反应条件下,作者首先对一系列炔醇醚底物的应用范围进行考察 (Scheme 2)。研究表明,上述的标准反应条件对于末端无取代的炔醇醚底物,均能够良好地兼容,并获得较高收率的环加成产物3a与3b。然而,对于O-苯基中具有供电子基团取代的炔醇醚底物,则观察到反应收率出现显著降低 (3c与3d)。接下来,作者进一步对一系列烯基化合物的应用范围进行深入研究。该小组发现,上述的标准反应体系对于各类取代的α-甲基苯乙烯底物,均能够以良好的反应收率 (67-90%),获得预期的环加成产物3e–3g。然而,研究表明,增大α-取代基的立体位阻,则能够观察到环加成产物3h的收率出现显著降低。之后,作者发现,各类苯乙烯 (3k–3o)以及烯丙基三甲基硅烷 (3p–3r)底物,同样无法获得较高的反应收率。并且,作者进一步发现,对于未取代的炔醇醚与非环烯基底物之间的环加成过程,则仅有3ee能够获得良好的反应收率与区域选择性。而环庚烯、环辛烯以及1,5-环辛二烯底物,则获得相应的双环衍生物3s–3x。之后,作者发现,在上述的标准反应条件下,环己烯底物则表现出较低的反应活性,而(+)-蒈烯却能够获得预期的[2+2]环加成产物3y与3z。而在β-石竹烯 (β-caryophyllene)参与的[2+2]环加成反应中,则观察到反应能够区域选择性地在烯基底物中的三取代双键位置进行,并获得相应的目标产物3hh (57% 收率, 3:1 rr),同时,表现出高度的立体专一性。此外,研究发现,具有较高立体位阻的烯醇醚底物,同样能够与上述的标准反应条件良好地兼容,并分别以79%以及92%的反应收率,获得预期的取代环丁基烯醇醚产物3ii与3jj。同时,作者发现,在催化剂A存在的条件下,各类内炔醇醚1e–1h与脂肪族烯基化合物 (3kk–3nn,3rr,3ss)或非活化苯乙烯 (3oo,3pp)之间的[2+2]环加成过程,同样能够获得中等至优良反应收率的预期产物。值得注意的是,在环加成产物3qq中,则观察到相反的区域选择性。接下来,该小组进一步发现,富电子的苯乙烯底物与内炔醇醚之间,则优先进行相应的形式[4+2]环加成过程,进而获得各类双环产物4a–4f。
接下来,作者进一步通过酸性条件下 (p-TSA•H2O或HCl)的水解过程,将上述的环烯醇醚产物转化为相应的环丁酮产物5a–5e (41-75% 收率) (Scheme 3A)。同时,研究发现,在Br2或NCS存在时,上述的烯醇醚产物3ab以及3af则能够通过α-卤化以及后续的水解过程,转化为相应的α-卤代环丁酮产物6a、6b以及6c (Scheme 3B)。

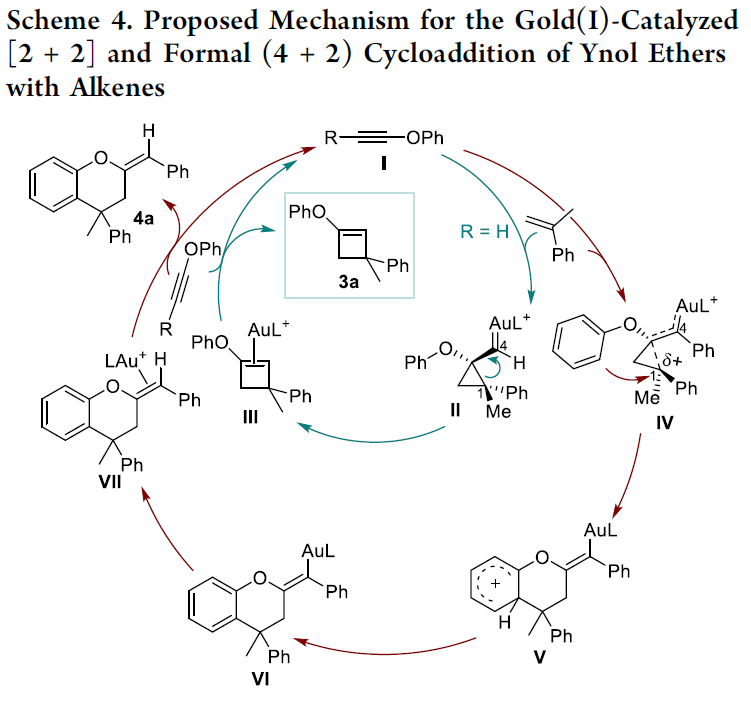
接下来,基于DFT计算,作者提出一种合理的反应机理 (Scheme 4)。首先,通过烯基化合物对于金(I)活化的炔基醚I的进攻,形成环丙基金(I)卡宾中间体II与IV。之后,通过中间体II的扩环过程,形成中间体III,并通过配体取代过程,形成相应的环烯醇醚产物3a。相反地,苯基取代的环丙基金卡宾中间体IV的结构则更加类似于通过高烯丙基金(I)稳定化的碳正离子。接下来,中间体IV经历后续的Friedel-Crafts过程,优先形成相应的Wheland中间体V。再通过V的去质子化过程,转化为中间体VI,并进一步经历后续的质子去金化 (protodeauration)以及配体交换步骤,最终获得相应的环丁烯酮产物4a。其中,区分上述两种不同机理路径的关键则在于烯键C1位置中相关取代基的稳定化作用。
总结
西班牙ICIQ的A. M. Echavarren课题组成功设计出一种采用金催化剂促进的芳基炔醇醚与烯基化合物之间的区域选择性[2+2]环加成反应方法学,进而成功完成一系列环丁酮分子的构建。同时,作者进一步发现,内炔醇醚与各类富电子的苯乙烯底物之间,则优先进行相应的形式[4+2]环加成过程。
参考文献
[1] B. B. Snider, Chem. Rev. 1988, 88, 793. doi: 10.1021/cr00087a005. [2] Y. Wang, Z. Zheng, L. Zhang, Angew. Chem. Int. Ed. 2014, 53, 9572. doi: 10.1002/anie.201403796. [3] B. M. Trost, J. Xie, N. Maulide, J. Am. Chem. Soc. 2008, 130, 17258. doi: 10.1021/ja807894t. [4] Y. Deng, L. A. Massey, P. Y. Zavalij, M. P. Doyle, Angew. Chem. Int. Ed. 2017, 56, 7479. doi: 10.1002/anie.201704069. [5] Z. Zheng, Y. Wang, X. Ma, Y. Li, L. Zhang, Angew. Chem. Int. Ed. 2020, 59, 17398. doi: 10.1002/anie.202003698. [6] J. Xie, P. Xu, Y. Zhu, J. Wang, W. C. C. Lee, X. P. Zhang, J. Am. Chem. Soc. 2021, 143, 11670. doi: 10.1021/jacs.1c04968. [7] J. Koudelka, T. Tobrman, Eur. J. Org. Chem. 2021, 2021, 3260. doi: 10.1002/ejoc.202100464. [8] (a) F. Marion, J. Coulomb, C. Courillon, L. Fensterbank, M. Malacria, Org. Lett. 2004, 6, 1509. doi: 10.1021/ol049530g.(b) S. Couty, C. Meyer, J. Cossy, Angew. Chem. Int. Ed. 2006, 45, 6726. doi: 10.1002/anie.200602270.
[9] R. B. Dateer, B. S. Shaibu, R. S. Liu, Angew. Chem. Int. Ed. 2012, 51, 113. doi: 10.1002/anie.201105921. [10] R. F. Sweis, M. P. Schramm, S. A. Kozmin, J. Am. Chem. Soc. 2004, 126, 7442. doi: 10.1021/ja048251l. [11] V. López-Carrillo, A. M. Echavarren, J. Am. Chem. Soc. 2010, 132, 9292. doi: 10.1021/ja104177w. [12] Y. B. Bai, Z. Luo, Y. Wang, J. M. Gao, L. Zhang, J. Am. Chem. Soc. 2018, 140, 5860. doi: 10.1021/jacs.8b01813. [13] M. Kreuzahler, G. Haberhauer, J. Org. Chem. 2019, 84, 8210. doi: 10.1021/acs.joc.9b01371. [14] M. E. de Orbe, M. Zanini, O. Quinonero, A. M. Echavarren, ACS Catal. 2019, 9, 7817. doi: 10.1021/acscatal.9b02314.
















No comments yet.