
作者:石油醚
引言
FDA近期批准了多款放射性药物,标志着精准肿瘤学进入新阶段,进而重塑癌症管理的临床实践。PET是现代癌症管理的核心工具,其可提供解剖成像无法获取的功能与分子信息。小分子(如[18F]FDG)可用直接亲核取代法合成;但大分子需借助前体基团,且其传统合成步骤繁琐、收率低。过去20年,基于硼酸盐/硅酸盐、硅烷、磷酸盐和Al18F螯合物的单步氟化策略大幅简化流程;近年更拓展至磷、硫等新型受体。
自2014年起,科学家聚焦两性离子有机三氟硼酸盐(AMBF3)前体,利用19F/18F同位素交换反应,实现复杂生物分子的“即装即用”式、一步、后期18F标记。AMBF3 prosthetic 基团成功用于多种分子支架的18F标记。目前主流偶联策略为CuAAC反应。少数情况下,药效团自带叔胺可经ICH2Bpin直接N-烷基化季铵化Mol. Pharmaceutics 2021, 18 (1), 187−197,再转化为AMBF3结构。尽管CuAAC成熟可靠,但因其铜离子可能引发生物分子氧化损伤,学界正积极推动羧基端AMBF3参与的酰胺键偶联策略,以实现无铜、更温和的肽标记。
近期,研究发现,AMBF3与靶向肽之间的连接化学结构直接影响体内性能。在CXCR4靶向肽示踪剂的头对头比较中,不含三唑环、仅含酰胺键的18F-BL08(图1A)展现出更优的肿瘤/肾脏比值;而结构高度相似但含CuAAC衍生1,4-三唑键的18F-BL09(图1B)虽肿瘤摄取相近,肾脏滞留却高达7.5%ID/g。其中,18F-BL08的肾脏摄取较18F-BL09降低3.4倍,显著改善肿瘤/肾脏对比度,证实AMBF3-Abu-OK(1)这一酰胺型连接体对体内药代动力学具有关键调控作用。
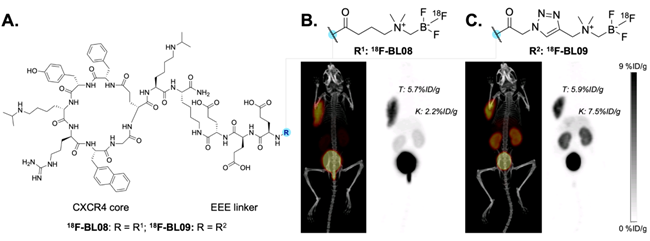
图 1 活性对比
AMBF3‑Abu-OK的合成路线
[路线一][ Mol. Pharmaceutics 2021, 18 (1), 187−197]
该路线存在(Scheme 1A)存在明显瓶颈:以GABA(5)为起始,经Eschweiler–Clarke甲基化和Fischer酯化得苄酯(6),两步转化均不完全,且叔胺6的柱层析纯化繁琐(收率仅52%);后续涉及保护基切换、AgNO₃/Pd催化、以及使用强腐蚀性TFA和剧毒KHF₂构建BF₃基团,操作危险、步骤冗长;更关键的是,中间体7在硅胶柱上极易脱氟分解,必须极速过柱,导致批次间脱保护效率波动大、产物交付不可控,单批产量长期受限于<500 mg(图2)。
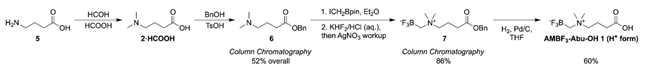
图 2路线一
[路线二][ Org. Process Res. Dev. 2026][10 g规模]
以2·HCl为起始,一步Boc保护(Boc₂O)与原位烷基化(ICH₂Bpin)直接生成4·I⁻;经Na₂S₂O₃水溶液淬灭及AG 1-X8树脂阴离子交换,高效获得33 g(92 mmol,63%总收率)的4·Cl⁻。随后在KHF₂/H₃PO₄中一锅完成氟化与叔丁酯脱除,直接得到10.8 g(32.6 mmol,42%)目标构建单元1(以2·HCl计收率26%)。该路线将线性步骤由5步压缩至2步,彻底取消两次硅胶柱纯化(避免叔胺6与三氟硼酸盐7的分离难题),剔除AgNO₃、Pd/C和TFA三种高危/高成本试剂,并实现脱保护与氟化“一锅化”,显著提升工艺稳健性与可放大性(图3)
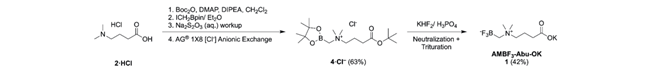
图 3路线二



















No comments yet.