
作者:石油醚
导读:
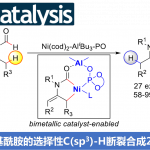
近日,北京理工大学夏中华课题组在iScience期刊中发表论文,报道一种新的芳基硼酸和末端芳基炔氧化Sonogashira交叉偶联反应策略,进而成功实现了一系列二芳基炔分子的构建。该反应策略经历的起始步骤,可能涉及到碱辅助的一价金(I)催化剂和芳基硼酸之间的转金属化过程。
Base-assisted transmetalation enables gold-catalyzed oxidative Sonogashira coupling reaction
L. Zhang, W. Zhang, D. Li, R. Yang, Z. Xia,
iScience. 2023, 27, 108531. doi: 10.1016/j.isci.2023.108531.
正文:
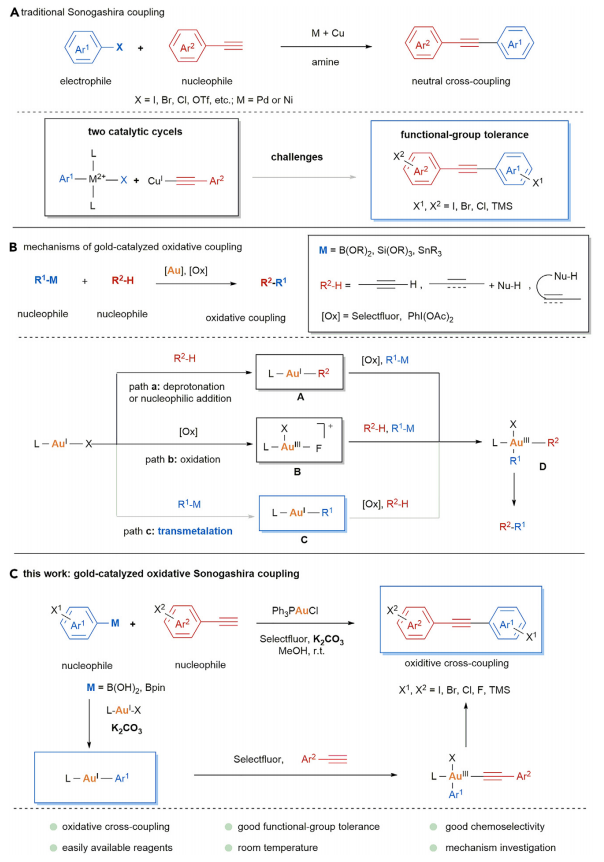
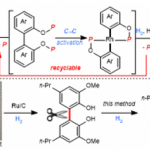
二芳基炔广泛存在于药物分子和有机材料中。芳基卤化物和末端炔之间的Sonogashira反应是合成二芳基炔最常用的偶联反应之一 (Figure 1A)[1]。对于金催化构建C-C键偶联反应方法学,目前仍有待进一步的研究报道。金催化氧化交叉偶联反应的两种常见的起始步骤,第一种是从末端炔的去质子化作用或亲核加成开始,形成功能化的一价金(I)中间体;第二种是一价金(I)在外部氧化剂的作用下形成三价金(III) (Figure 1B)[2]-[3]。近日,北京理工大学夏中华课题组报道一种新的碱辅助的金(I)催化剂和芳基硼酸之间的转金属化,进而和末端芳基炔发生氧化Sonogashira交叉偶联反应,成功实现了一系列二芳基炔分子的构建 (Figure 1C)。
![]()
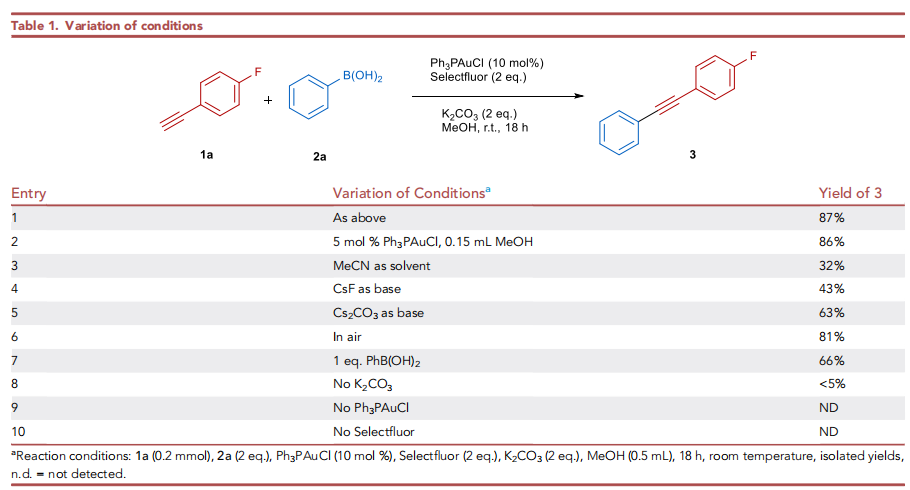
首先,作者采用对氟苯乙炔1a与苯硼酸2a作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用Ph3PAuCl作为催化剂,Selectfluor作为氧化剂,K2CO3作为碱,MeOH作为反应溶剂,反应温度为室温,最终获得87%收率的二芳炔产物3。
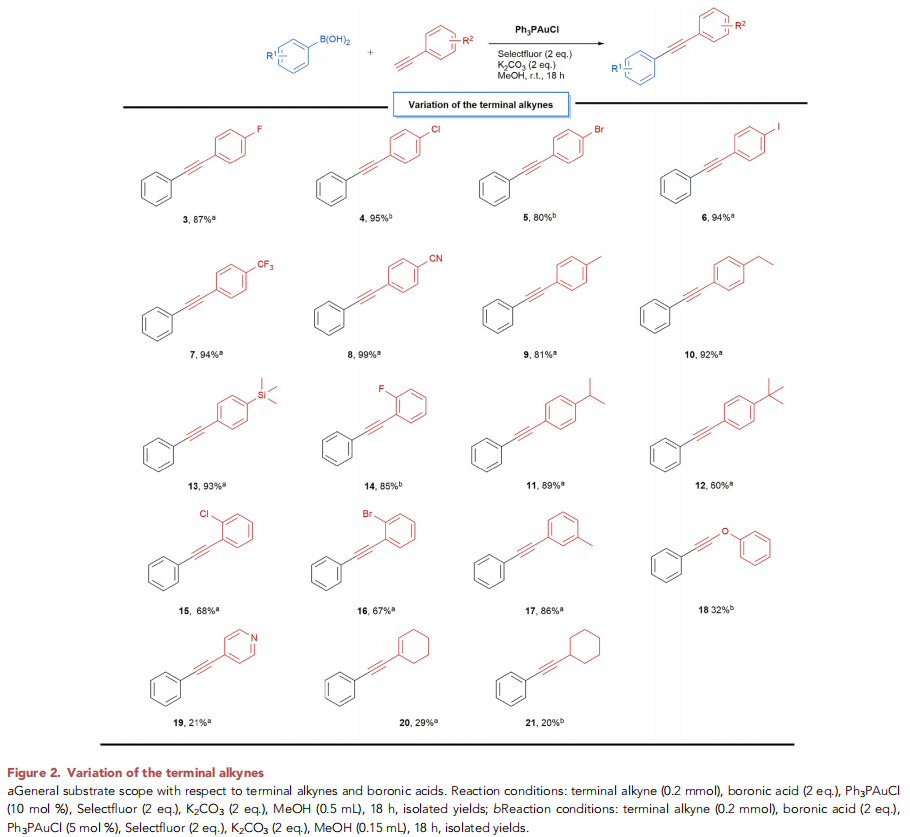
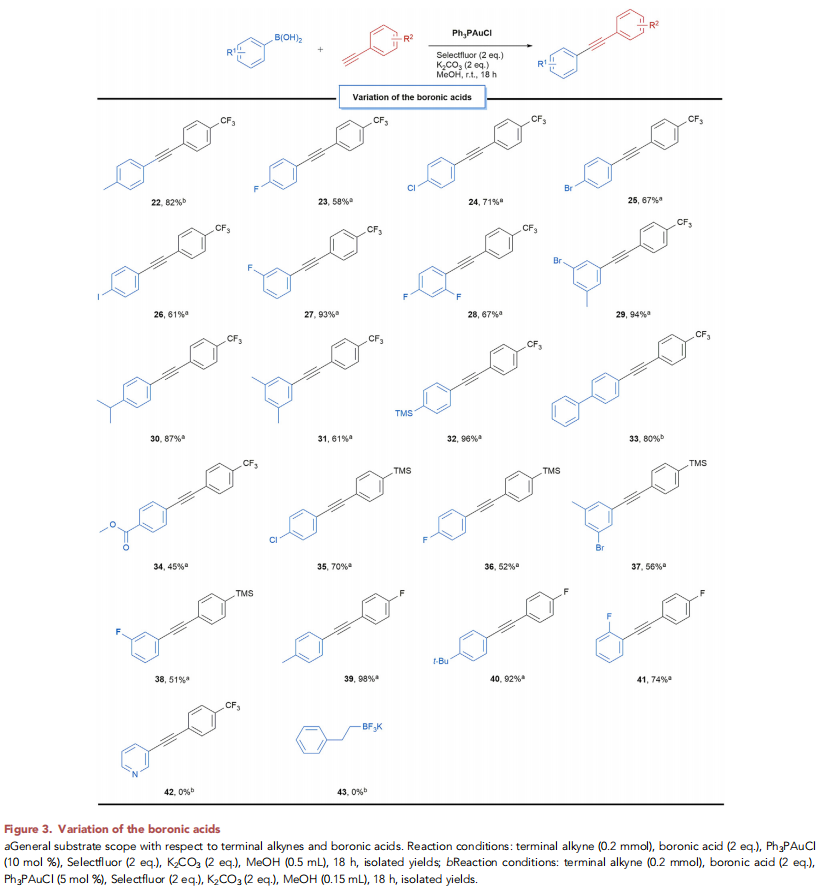
在上述的最佳反应条件下,作者分别对一系列末端炔底物 (Figure 2)以及硼酸底物 (Figure 3)的应用范围进行深入研究。


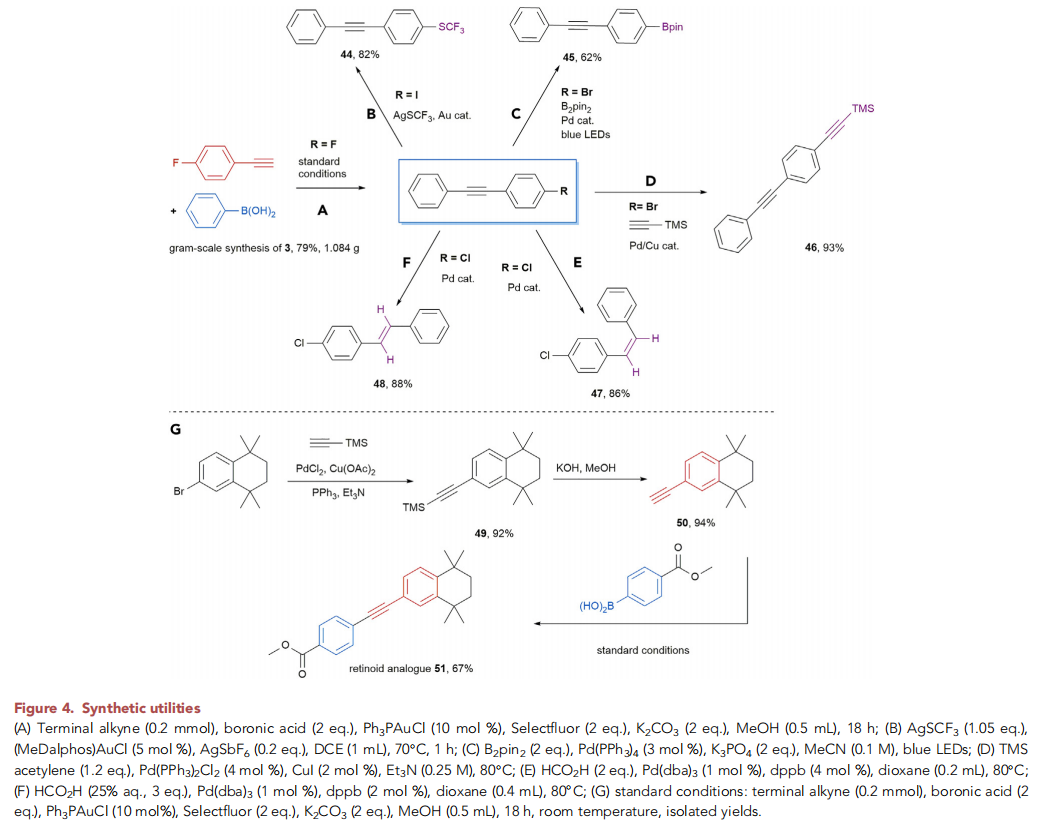
之后,该小组通过如下的一系列研究进一步表明,这一新的交叉偶联策略具有潜在的合成应用价值 (Figure 4)。
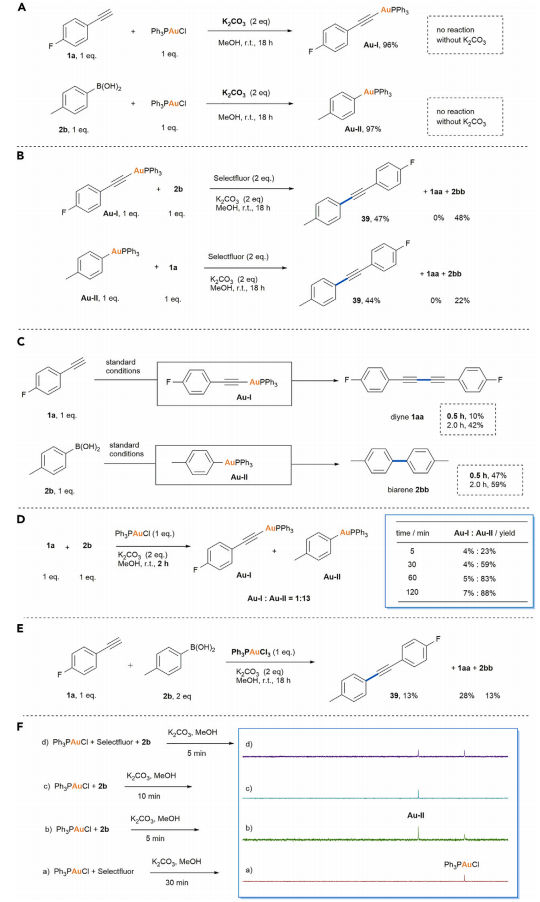
接下来,作者对上述偶联过程的反应机理进行了研究 (Figure 6)。


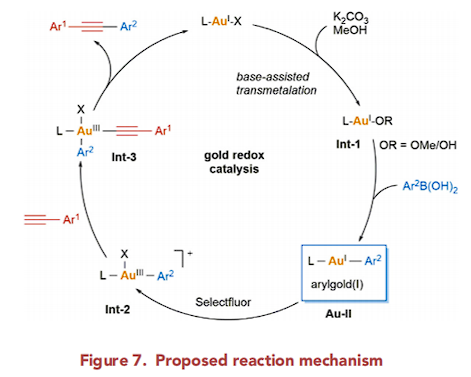
基于上述的实验研究以及前期相关的文献报道[4],作者提出如下可能的反应机理 (Figure 7)。
总结:北京理工大学夏中华课题组报道一种新的芳基硼酸和末端芳基炔氧化Sonogashira交叉偶联反应策略,进而成功实现了一系列二芳基炔分子的构建。这一新的二芳炔合成策略具有操作简单、反应条件温和、底物范围广泛以及优良的官能团兼容性等优势。
参考文献:
- [1] R. Chinchilla, C. Nájera, Chem. Rev. 2014, 114, 1783. doi:10.1021/cr400133p.
- [2] A. Leyva-Pérez, A. Doménech-Carbó, A. Corma, Nat. Commun. 2015, 6, 6703. doi:10.1038/ncomms7703.
- [3] K. Liu, N. Li, Y. Ning, C. Zhu, J. Xie, Chem. 2019, 5, 2718. doi:10.1016/j.chempr.2019.07.023.
- [4] D. V. Partyka, M. Zeller, A. D. Hunter, Angew. Chem. Int. Ed. 2006, 45, 8188. doi:10.1002/anie.200603350.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.













No comments yet.