
本文作者:石油醚
导读
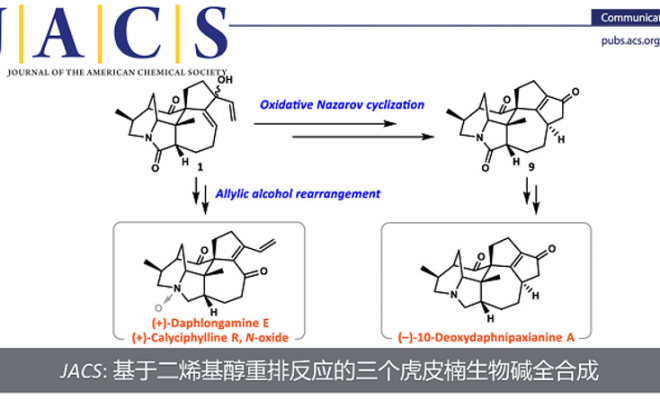
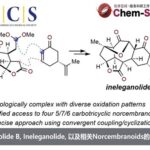
近日,南方科技大学理学院化学系徐晶教授课题组在对Calyciphylline A-型虎皮楠生物碱的相关合成工作中,发现了一种新颖的基于二烯基醇结构的氧化重排反应,并借此完成了虎皮楠生物碱(−)-10-Deoxydaphnipaxianine A的首次不对称全合成。另外,通过改变溶剂调控该反应的选择性发生正常的烯丙醇重排反应,完成了(+)-Daphlongamine E 和 (+)-Calyciphylline R的首次不对称全合成。南方科技大学理学院化学系徐晶教授是本文的通讯作者;南方科技大学理学院化学系博士生张焱和访问学者陈雨叶为共同第一作者。
“Total Syntheses of Calyciphylline A-Type Alkaloids (−)-10-Deoxydaphnipaxianine A, (+)-Daphlongamine E and (+)-Calyciphylline R via Late-Stage Divinyl Carbinol Rearrangements
Yan Zhang, Yuye Chen, Manrong Song, Bin Tan, Yujia Jiang, Chongyuan Yan, Yuyang Jiang, Xinyue Hu, Chengqian Zhang, Wenqing Chen, and Jing Xu*
J. Am. Chem. Soc., 2022, ASAP, doi: 10.1021/jacs.2c05957
正文
虎皮楠生物碱是从虎皮楠科植物中分离出来的一大类生物碱,它们几乎都具有多环笼状骨架,具有非常大的合成挑战性。因此也深受很多合成化学家的青睐。自Heathcock教授经典的仿生合成路线报道出来之后,近年来,Carreira, 李昂,Smith,Fukuyama/ Yokoshima, Dixon, 翟宏斌, 邱发洋, 高栓虎, Sarpong, 李超和陆海华等国内外课题组先后完成了几十个虎皮楠生物碱的全合成工作,其中Calyciphylline A-型虎皮楠生物碱的合成属于前沿热点之一。
徐晶教授课题组利用TEMPO类型的氧化剂的反应体系,通过调控反应的选择性,分别得到了两种不同的Calyciphylline A类型的虎皮楠生物碱骨架。并完成了三个虎皮楠生物碱的首次不对称全合成。
工作详细解析:关键反应的发现
作者在进行虎皮楠生物碱(+)-Daphlongamine E的相关合成工作过程中,在合成二烯基醇中间体1后,原本设想通过Dauben−Michno重排反应构建分子中的α,β,γ,δ-二烯酮结构,即化合物2。在查阅文献之后,面对化合物1这种非常复杂的底物结构,作者选择用Iwabuchi教授发展的TEMPO+BF4–参与的Dauben−Michno重排反应。在得到化合物1之后,作者发现在该条件下(TEMPO+BF4–, MeCN)顺利发生了反应,生成了一个在紫外灯下有较强荧光的产物。但是经过作者分离并通过核磁表征之后,发现核磁氢谱上没有任何烯基氢,核磁碳谱上虽然有两个酮羰基的峰,但是结合DEPT 135谱图来看只有在151 ppm和173 ppm左右有两个全取代的双键碳,除此之外并没有其他的双键碳。在通过高分辨质谱表征给出了与作者目标产物相同的分子量后,作者提出了底物发生环化后并氧化的猜想,即生成了化合物3,之后便通过单晶结构证实了这一点。这样就为含有符合化合物3这样的5-5环系提供了基础。
在作者得到这一结果之后,作者想对此反应进行优化,尝试是否能通过更换其他条件来使Dauben−Michno重排反应得以正常发生。令人惊喜的是,在作者尝试了以1,4-二氧六环作为溶剂时,作者发现中间体1发生了烯丙醇的重排反应得到了具有共轭二烯醇结构的化合物4。为符合化合物2结构的虎皮楠生物碱的合成提供了研究基础。
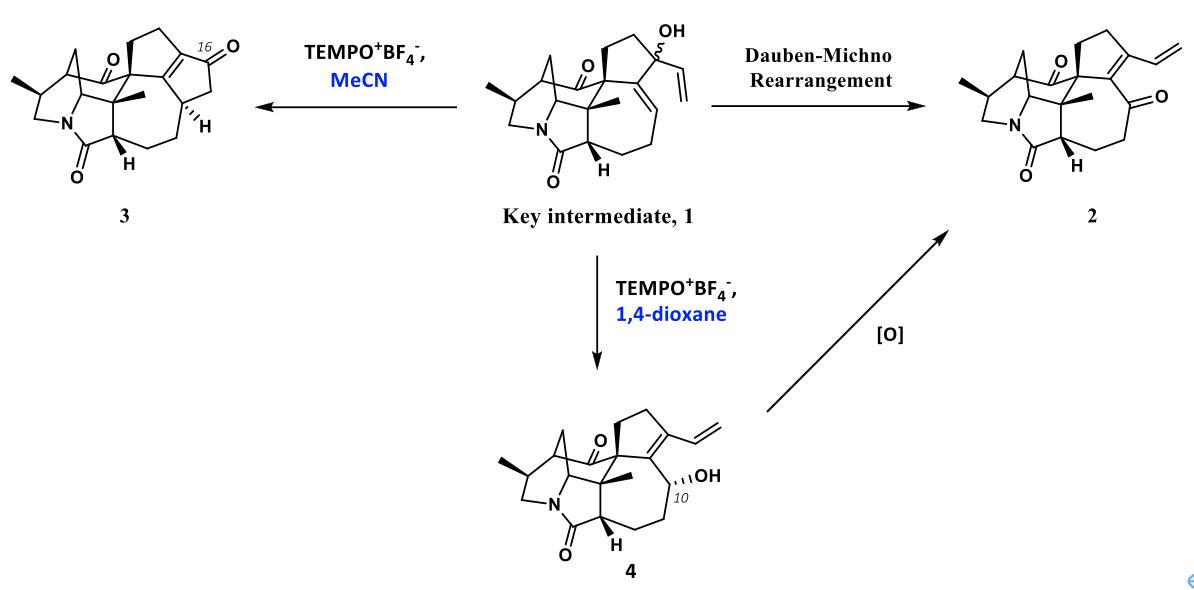
图1 前言介绍
目标分子的逆合成分析
最初对daphlongamine E进行了逆合成分析(如图2),二烯酮结构可以由重排反应得到,其二烯基醇前体1可由化合物5通过pinacol偶联以及格氏反应得到;化合物5可由化合物6通过Tsuji-Trost反应引入季碳之后再由硼氢化氧化反应引入氧化态;化合物6可由化合物7通过逐级氧化制备,化合物7可以通过Heck偶联等反应制备;化合物8可由化合物9通过水解氰基并发生Michael加成得到;化合物9可根据作者课题组之前的工作制备。
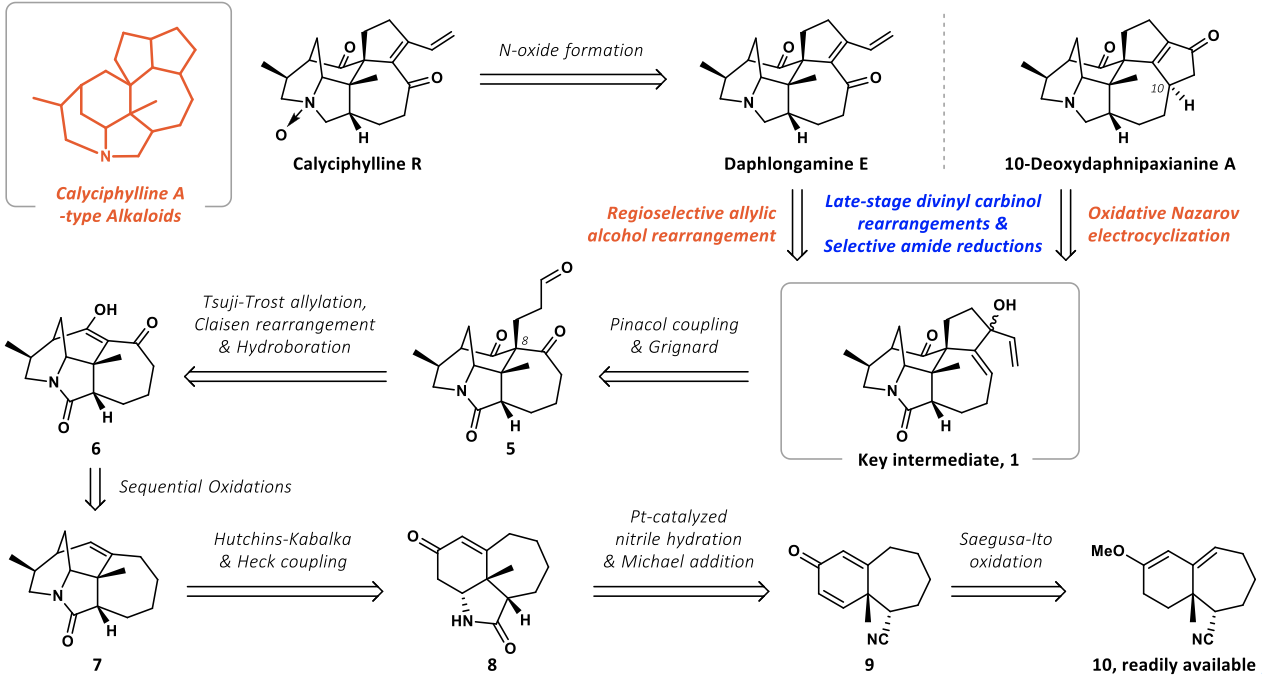
图2 逆合成分析
Deoxydaphnipaxianine A, (+)-Daphlongamine E 和 (+)-Calyciphylline R的不对称全合成
如图3所示,化合物10通过水解以及Saegusa氧化得到化合物9。化合物9通过使用Grubbs教授课题组发展的高效氰基水解催化剂来水解氰基并发生分子内的aza-Michael反应得到化合物8。化合物8与对甲苯磺酰肼缩合后与儿茶酚硼烷发生作用发生Hutchins-Kabalka重排反应,并通过与2,3-二溴丙烯反应引入侧链并通过Heck偶联反应关环及Rh催化剂的催化氢化得到化合物7。化合物7在SeO2作用下发生烯丙位氧化后,再通过B2Pin2的共轭加成及氧化得到化合物6。化合物6通过Tsuji-Trost反应构建季碳并通过Wilkinson催化剂催化的硼氢化氧化反应引入氧化态,进而通过PCC氧化得到化合物5。化合物5在SmI2介导下发生区域选择性的Pincaol偶联反应关环,通过Ley氧化并消除羟基得到烯酮,进而由乙烯基格氏试剂进攻得到关键中间体1。中间体1以乙腈为溶剂在TEMPO+BF4–的作用下发生了氧化Nazarov环化反应得到化合物3。但是经过尝试,很多条件都无法完成最后的选择性的酰胺还原,最后决定使用比较稳定的肟基团作为保护基,并通过Lawesson反应得到硫代酰胺最后在雷尼镍的作用下脱硫得到了10-Deoxydaphnipaxianine A。另外中间体1以1,4-二氧六环做溶剂,在TEMPO+BF4–的作用下发生了烯丙位重排反应得到了化合物4,推测是由于重排后羟基位阻非常大,所以并没有继续被氧化成酮。于是作者将其分离出来后,使用了位阻较小的AZADOL/PIDA的氧化体系成功得到了化合物2。最后通过一步Vaska催化剂催化的选择性酰胺还原得到(+)-Daphlongamine E,再通过m-CPBA氧化得到(+)-Calyciphylline R。
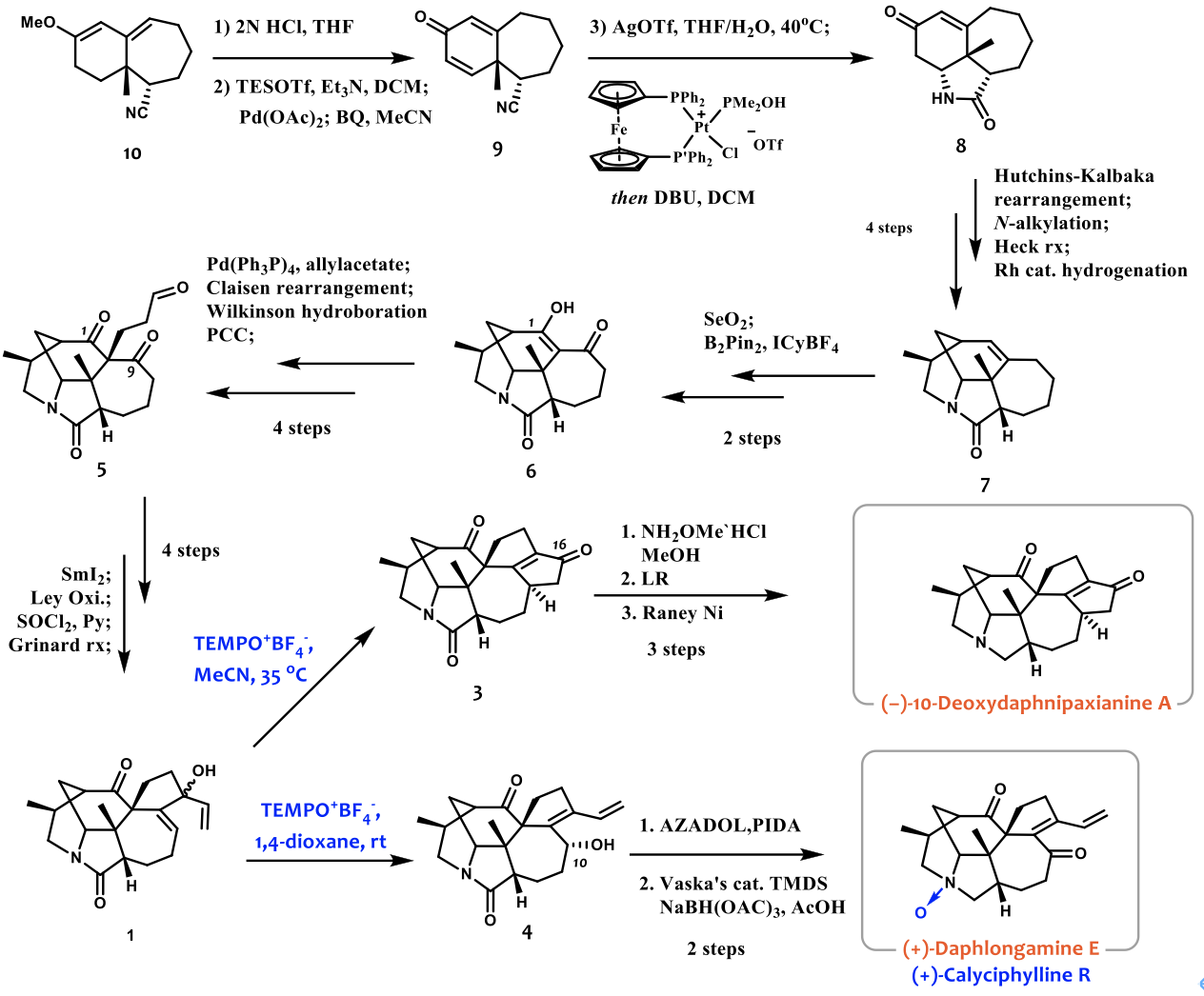
图3 全合成路线
氧化Nazarov环化反应的机理
关于这个转化,作者提出了可能的机理(如图4)。首先中间体1的羟基对TEMPO+BF4-进行加成,之后离去基团离去后形成戊二烯正离子并迅速发生4π电环化反应关环,之后被氧负离子捕获后氧化,最终得到化合物3。
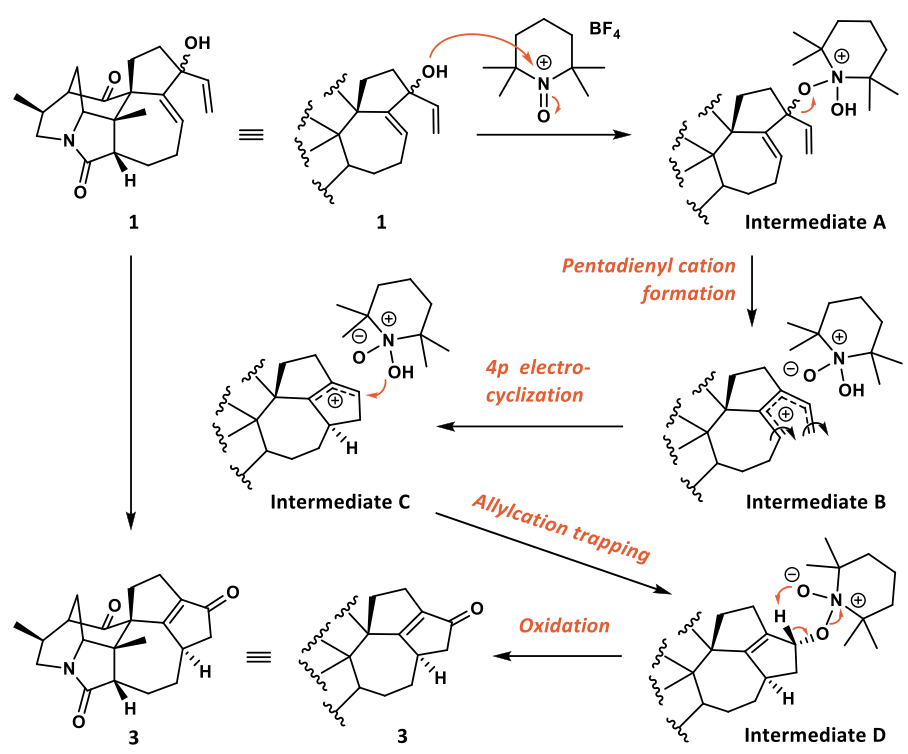
图 4 氧化Nazarov环化反应的机理
总结与展望
徐晶教授课题组通过对未按预期生成的产物进行分析发现了一个新颖的氧化Nazarov电环化反应,并以此为关键反应完成了(−)-10-Deoxydaphnipaxianine的首次不对称全合成。另外通过对该反应的溶剂更换实现了选择性调控发生烯丙醇重排反应,并以此完成了虎皮楠生物碱(+)-Daphlongamine E 和 (+)-Calyciphylline R的首次不对称全合成。
(徐晶教授供稿)












No comments yet.