

本文作者 Cyclization Xu
西安交通大学前沿科学技术研究院的李鹏飞教授课题组长期致力于有机硼化学和吡啶衍生配体的研究。在配体设计领域,李鹏飞课题组开发了双吡啶取代的联硼化合物和吡啶取代的硼硅化合物作为N,B-双齿配体的前配体,并应用其高效地实现了铱催化芳基C-H硼化反应,开拓了硼基配体的新领域。近期,该课题组开发了一类结构高度可调节的新骨架手性吡啶单元,将其引入N,B-配体实现了铱催化不对称C-H硼化反应,并开发了新型手性联吡啶配体并成功应用于镍催化还原偶联反应。李鹏飞教授课题组吡啶衍生配体的研究丰富了过渡金属催化反应的基础理论,也为面向药物和材料的有机合成提供了新手段。
下面将对李鹏飞教授课题组开发的系列吡啶衍生配体的发展做以梳理与介绍(图1)。
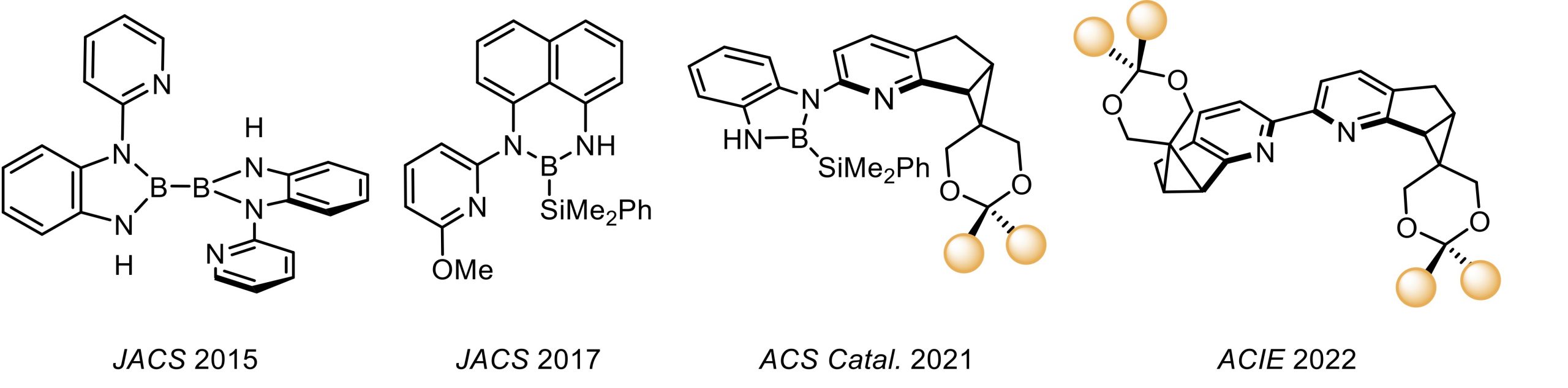
图1. 李鹏飞课题组设计的系列吡啶衍生配体
1. N,B–双齿硼基配体与铱催化C–H硼化反应
在过渡金属催化反应中,配体对催化剂活性与选择性的调控具有重要意义。在常见配体中,配位原子主要包括磷、氮、碳、氧等,例如,氮杂环卡宾配体的配位原子为带有六个电子的sp2碳原子。sp2杂化的硼负离子与卡宾互为等电子体,且电子构型一般同为单线态。然而,以硼原子作为配位原子的配体在催化反应中极为少见。与碳相比,硼元素电负性较低,sp2杂化的硼负离子具有很强给电子能力,并且具有一个空的p轨道。因此,硼负离子具有更强的σ给电子能力和可调节的π吸电子能力(图2)。
将硼基配体用于催化反应需克服两个困难:第一,目前缺少有效的方法将硼基方便地引入金属中心;第二,硼-金属键的高反应性使其一般仅作为“反应”配体经金属转移到产物中,而无法“支持”配体进行催化循环。
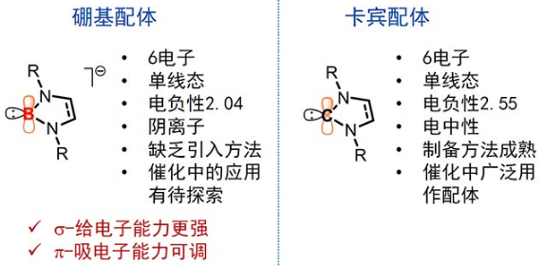
图2. 硼基配体与氮杂环卡宾配体的比较
过渡金属催化C-H硼化反应是构建C-B键的有力途径,有机硼化合物是有机合成、药物研发和材料发展中广泛使用的重要中间体。在铱催化C-H硼化反应中,2,2′-联吡啶和1,10-菲咯啉是广泛使用的高活性配体,催化机理如图3所示。其中,具有三个硼基的IrIII配合物中间体是其活性催化剂,经过与芳基C-H键发生氧化加成和还原消除得到目标产物,而氧化加成是催化循环的决速步。值得一提的是,在活性催化剂中,仅有一个硼基能够转化为产物,另外两个硼基仅作为配体的角色存在,因而为调控催化剂电子特征与空间位阻提供了机会。通过强给电子配体提高中心金属的电子云密度,能够有效的促进氧化加成的发生,从而提高反应的催化活性。
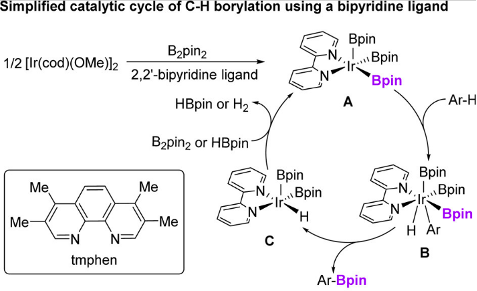
图3. 铱催化碳氢硼化反应的催化循环
受铱催化的硼化反应机理与氮杂环卡宾配体的启发,李鹏飞课题通过对催化反应中间体的联吡啶和硼基进行了巧妙重组,设计了具有更强给电子能力的N,B-双齿配体。通过独创的双吡啶取代联硼前体化合物,利用吡啶辅助的B-B键氧化加成反应,将两个N,B-双齿配体引入金属铱,并通过晶体结构证明了该方法的有效性(图4)。
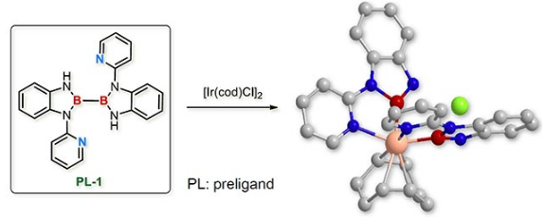
图4. 双吡啶取代的联硼化合物及其氧化加成
他们进一步通过深入分析空间位阻决定选择性C-H键硼化反应的机制,发展了具有双N,B-配体的铱催化剂。以此新催化体系在惰性碳氢键直接硼化的反应中获得当时最高的催化活性,并具有广泛的底物普适性(图5)。与文献催化剂比较,该催化体系对挑战性底物具有更好反应效果。
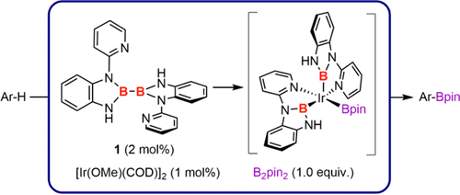
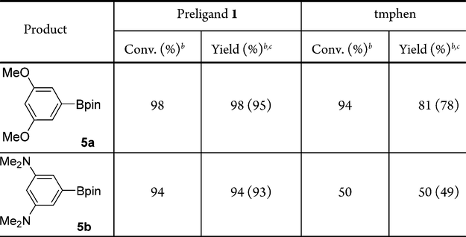
图5. N,B-双齿硼配体与空间位阻决定选择性的铱催化C–H硼化反应
该研究合成了具有双吡啶取代的联硼化合物,并将其作为N,B-双齿配体前体应用于铱催化芳烃硼化反应中。反应首次成功使用双齿硼负离子作为均相催化中的支持配体,并获得优异的催化活性,从而反映了硼配体的独特性,并为今后发展相关的配体和催化反应提供了新思路。
2. 新型N,B–双齿硼基配体前体与铱催化C–H硼化反应
通过设计独特的双吡啶取代的联硼前体化合物,利用吡啶辅助的B-B键氧化加成反应,李鹏飞课题组将两个N,B-双齿配体引入金属铱,发展了双N,B-配体的铱催化剂,并将其应用于空间位阻决定选择性的芳基C-H键硼化反应。然而,催化剂中的两个N,B-配体占据了IrIII的配位点,仅留有一个空轨道用于催化反应的进行。对于具有导向基的芳基C-H键硼化反应而言,反应需要有两个空的配位点才能使反应顺利进行,其一与底物中的导向基进行配位,其二实现芳基的碳氢活化(图6)。因此,基于双吡啶取代联硼化合物的第一代N,B-双齿配体具有局限性,需要进一步改进以适应不同的催化反应需求。
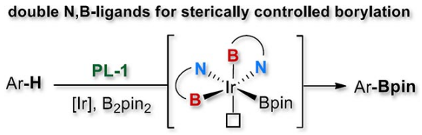
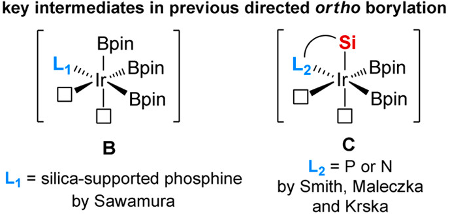
图6. 铱催化邻位导向方剂碳氢硼化反应中的配体设计
在前期研究基础上,李鹏飞课题组设计了硅硼烷作为配体前体,通过B-Si键的氧化加成将单个N,B-配体引入过渡金属。选择硅硼烷作为N,B-配体前体的原因如下:1)Si−B键能够和低价金属发生氧化加成反应;2)还原性硅基能够通过还原消除或配体交换选择性地从中心金属除去,从而释放金属的空轨道;3)大位阻硅基取代基能够抑制双N,B-配体铱配合物的生成。
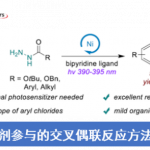
以原创的硅硼烷作为新的N,B-配体前体,李鹏飞课题组设计并成功发展了一种用于官能团导向的邻位C-H键硼化反应的催化剂。相对于文献报道的导向C-H键硼化反应,该催化剂不限于特定的导向基团,可以用于含酯、酰胺、氨基甲酸酯等常见官能团底物的直接硼化,应用范围较广且催化体系简单,收率较高(图7)。令人惊喜的是,该催化体系能够实现吡啶作为导向基的C(sp3)-H硼化反应,并取得了较高的产率。
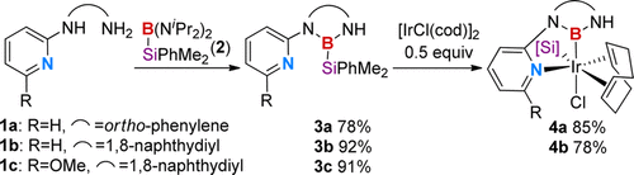
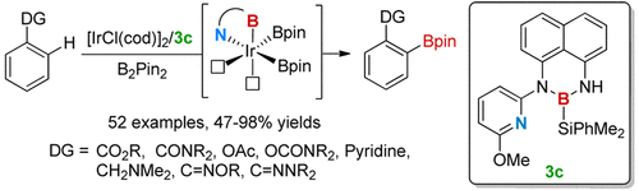
图7. N,B-双齿硼基配体参与的铱催化C-H键硼化反应
3. 手性N,B–双齿硼基配体前体与铱催化去对称化C–H硼化反应
李鹏飞课题组在催化领域首次提出支持型N,B-双齿配体,并利用其开发了迄今为止芳烃C-H硼化反应活性最高的催化剂之一,适用于多种类型底物的官能团导向邻位C-H 硼化反应。面向不对称金属催化反应的目标,手性配体的开发是关键所在,在调控催化剂活性和反应立体选择性中具有关键性作用。N,B-双齿配体的作为吡啶衍生配体,其手性版本的开发的关键在于设计并合成手性吡啶单元。
吡啶是催化中应用最广泛的配体结构单元,但至今缺乏普适性的手性吡啶骨架,制约了众多不对称催化反应的发展。半个世纪以来,许多化学家尝试发展手性吡啶配体(图8),但在同时解决三方面主要挑战中遇到困难:第一,向吡啶平面引入手性元素;第二,在骨架上方便地进行结构改造;第三,在吡啶环旁边引入取代基时,立体选择性的提高常常伴随催化活性的降低。这些困难激发了化学家极具创意的配体设计思想。因此,通过结构设计针对性地同时克服上述困难,建立一类结构高度可调节的手性吡啶结构单元对手性吡啶类的发展具有重要意义。
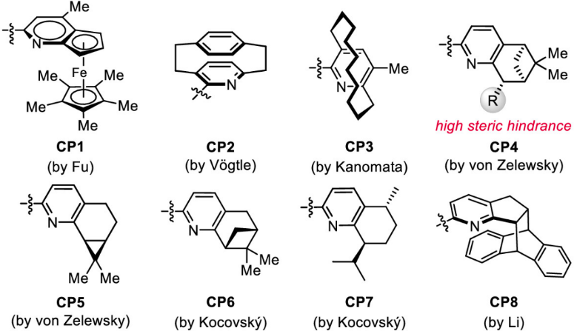
图8. 文献已报道的手性吡啶结构单元
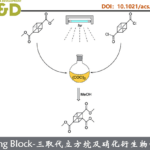
针对上述问题,李鹏飞课题组设计了一种结构可调新手性吡啶骨架。首先,采用稠环骨架减少配体可能的空间构象,增加其在不对称催化中的可预测性;第二,相比于常见的在吡啶2-位引入甲基等取代基,在吡啶并五元环骨架的基础上再稠合一个三元环能够有效减小吡啶氮原子周围的空间位阻,从而提高配体的反应活性;第三,相较于已有的手性吡啶配体,在环丙烷上引入羟甲基,实现了配体结构的高度可调性。为了验证新手性吡啶骨架在不对称催化中的效果,作者进一步合成了一系列手性吡啶衍生的N,B-配体(图9)。
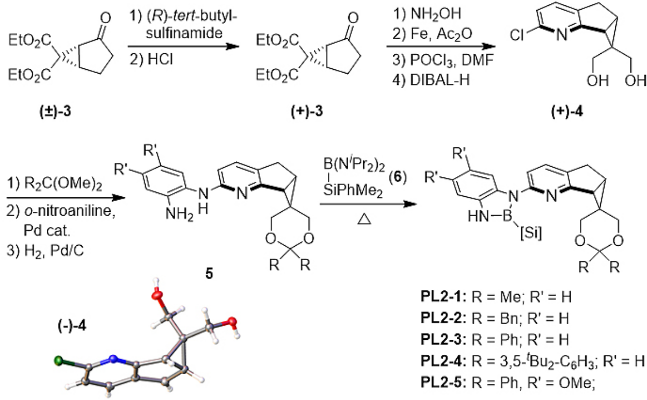
图9. 新骨架手性吡啶衍生的N,B配体的合成
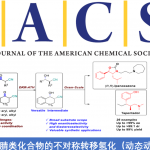
在获得新型手性吡啶衍生的N,B-配体之后,作者将其应用于不对称C-H键硼化反应中,以高对映选择性实现了三芳基甲烷的不对称C-H键硼化(图10)。为了更加清楚地认识手性配体对于产物对映选择性的贡献,作者对反应的决速步骤(C-H活化阶段)进行了理论计算。计算结果表明,配体缩酮部分椅式构型的朝向,以及手性配体和底物之间的氢键和C-H/π等弱相互作用,对于产物的对映选择性起到了关键性作用。
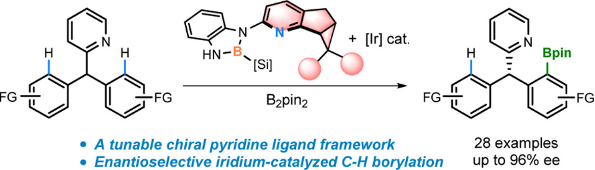
图10. 新骨架手性N,B配体促进的铱对映选择性C-H硼化反应
为了考察该催化反应的实用性和高效性,作者将反应的规模提高到克级,并减少了催化剂的用量(1 mol%),反应依然可以得到高对映选择性结果。产物的硼酸酯基团可以高效地转化为叠氮、溴、硒醚、二氟化硼等基团(图11)。产物经过氧化/氢化,可以顺利得到药物分子中常见的手性哌啶衍生物。
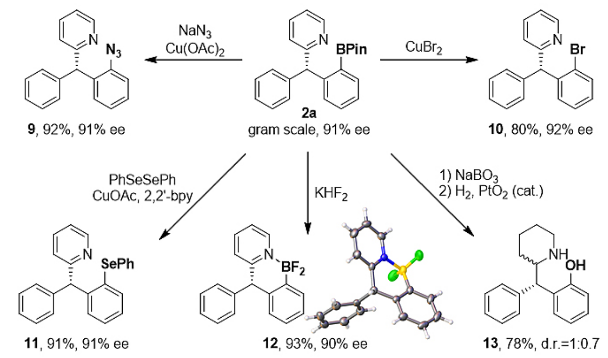
图11. 手性硼产物的实用性转化
该研究设计合成了一种结构可调新手性吡啶配体,并将其应用于不对称C-H硼化反应,高对映选择性地实现了三芳基甲烷的不对称C-H键硼化。更重要的是,该工作证明了这种新型手性吡啶配体的确可以在不对称催化反应中发挥关键性的作用。考虑到吡啶配体(如2,2’-联吡啶)在催化反应中的广泛用途,该手性吡啶骨架衍生的配体有望在不对称催化领域发挥更广泛的作用。
4. 新骨架手性联吡啶配体与不对称还原加成反应

2,2′-联吡啶配体是金属催化反应中广泛使用的经典配体,在廉价金属催化、仿酶催化及光、电催化等重要新兴领域常作为优选配体。然而,不对称催化领域至今仍然缺乏普适性的手性联吡啶配体,其主要困难是吡啶的芳香性平面结构难以引入手性元素,现有配体骨架可修饰性差,提高立体选择性常伴随催化活性降低。这些困难激发了化学家极具创意的配体设计思想(图12)。例如,大连化物所周永贵研究员发展了柔性链调控的轴手性联吡啶配体,并已在若干反应中取得优秀结果。另外,在有些反应中人们用噁唑啉或胺等替代吡啶,但常常导致活性打折,且难以满足众多反应的需求。因此,手性联吡啶配体的发展仍然是不对称催化领域亟待解决的重要挑战。
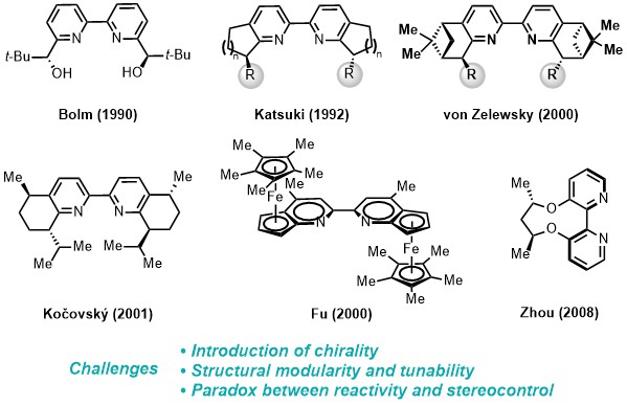
图12. 文献已报道的代表性手性联吡啶配体
分析上述手性联吡啶配体的结构特征,传统策略通常向2,2′-联吡啶的6和6′位置引入手性基团或者引入α位具有手性元素的5,6-和5′,6′-稠环结构(图13)。通过对典型结构的分析,以上两种方式都会在金属中心的近程区域引入较大的空间位阻,从而导致反应活性降低或者底物范围限制。
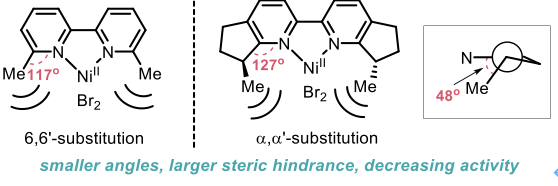
图13. 传统手性联吡啶配体的设计思路与局限性
酶催化反应通常对底物有高度的专一性,底物范围比较窄,但细胞色素家族却可以催化多种底物的氧化反应,并且其结构演化的类似物在近年来在酶催化中表现出优异的普适性。细胞色素的活性中心是具有大型平面共轭结构的铁卟啉配合物,卟啉是金属中心的“第一配位层”,在金属周围形成小位阻空旷区域,可以容纳各种形状的小分子。铁卟啉周围具有特定结构的蛋白质构成其“第二配位层”,形成了对底物结构具有一定选择性的手性微环境,存在着如氢键、离子相互作用、π-π堆积等弱相互作用力(图14)。这种“内松外紧”的特殊结构使得这些酶兼具高活性、高选择性和广泛适用性的优势。2018年诺贝尔奖得主Arnold教授已通过定向进化的手段,实现了包括不对称C-H键羟基化、不对称环丙烷化在内的许多反应。
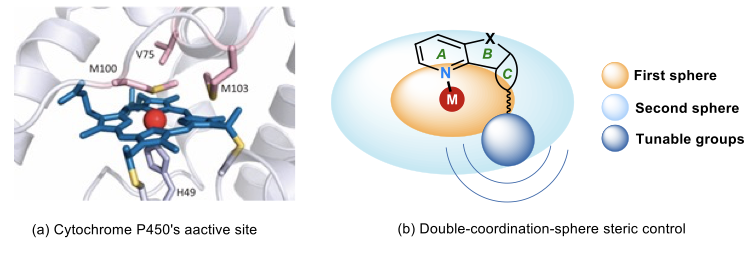
图14. 新型手性联吡啶配体的设计灵感来源
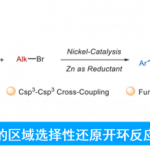
受酶结构启发,通过从头合成、预留修饰基团和结构设计,李鹏飞课题组针对性克服了手性吡啶配体的设计难题,建立了一类新骨架手性2,2′-联吡啶配体SBpy(图15)。根据键角和二面角的分析,该配体具有[6-5-3]刚性稠环结构,实现了中心金属近程位阻最小化和远端侧链结构的高度可调性,克服了立体选择性和催化活性之间的矛盾。有别于传统的手性配体设计思路,SBpy具有“内松外紧”的结构特征,与细胞色素P450异曲同工,为手性催化剂的设计提供了新思路。从原创的手性氯吡啶结构单元出发,经过镍催化还原偶联反应便可获得一系列手性联吡啶配体SBpy。
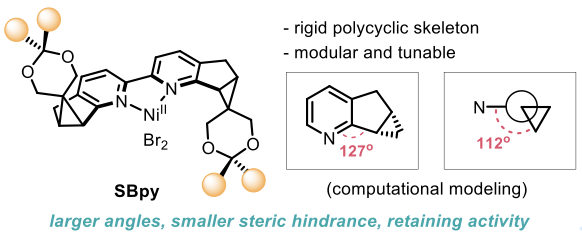
图15. 新骨架手性联吡啶的结构设计
作者通过药物合成中常见的羰基加成反应展示了SBpy设计的合理性与有效性(图16)。目前,当量金属试剂对羰基的不对称加成发展成熟,但以易得的卤化物为原料实现该转化则更具经济性和普适性。以二苯甲酮衍生的Ph-SBpy为配体,课题组首次实现了分子间芳基卤化物对芳基醛的不对称加成反应。该反应催化活性高,条件温和,大多数底物均取得了优秀的收率和对映选择性,成功应用于复杂药物的合成与衍生化,为手性分子的立体选择性合成提供了新工具。
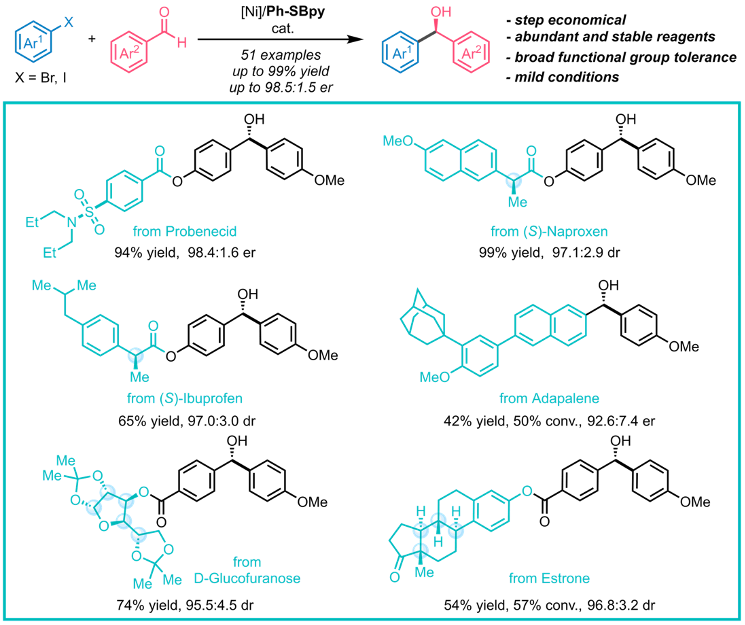
图16. Ph-SBpy促进的分子间不对称还原加成反应
作者进一步研究了配体结构与催化活性之间的关系(图17)。通过对一系列联吡啶配体进行比较,作者发现联吡啶上富电子取代基能显著提高催化活性,而近程位阻增大则会导致催化活性明显降低。在引入手性元素的同时,Ph-SBpy既具有富电子配体骨架,又使近程空间位阻最小化,还可通过侧链基团保证活性催化剂稳定性,因而即便在低温下仍然具有很高的催化活性。SBpy配体可以“活性不打折”地实现反应的立体选择性。
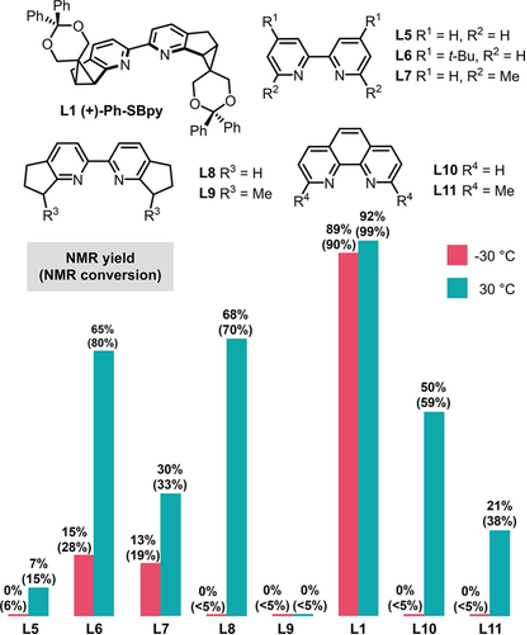
图17. Ph-Sbpy与其它联吡啶型配体的催化活性比较
作者通过机理实验提出镍催化芳基卤化物对醛不对称加成的可能机理。通过密度泛函理论计算,作者得到了立体选择性关键步骤的过渡态结构(图18)。在优势过渡态中,反应物苯甲醛的苯基和与临近的配体的苯基之间存在相互吸引的π–π堆积作用,进一步解释了Ph-SBpy比其他富电子、低位阻联吡啶配体活性更高的原因。
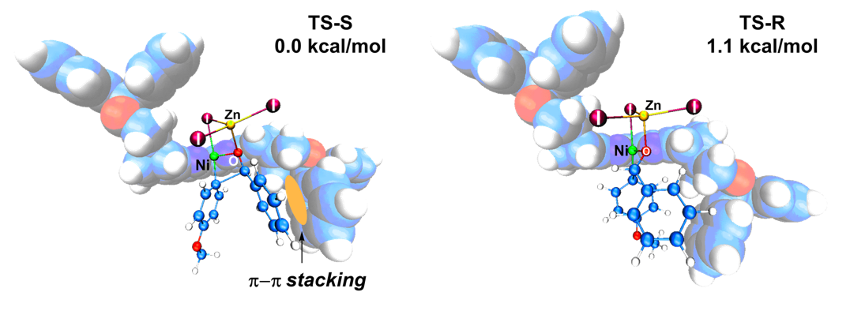
图18. Ph-Sbpy促进立体选择步骤的过渡态
总之,鉴于2,2’-联吡啶配体的广泛应用价值和手性版本发展受限的现状,李鹏飞课题组设计、发展了一类结构可调的新骨架手性联吡啶配体SBpy,并通过实现镍催化醛的不对称还原芳基化反应展示了新配体设计的有效性,该工作为手性分子的选择性合成提供了新工具。
希望李鹏飞教授课题组N,B-双齿配体和手性吡啶配体的设计与应用能够为相关催化剂和配体的设计提供新的思路。
研究者介绍

李鹏飞,西安交通大学前沿科学技术研究院教授、博士生导师,前沿基础科学中心主任,曾任前沿院科研副院长。2004年师从著名农药化学家和有机化学家南开大学李正名院士获得硕士学位,2010年博士毕业于德国海德堡大学,同年到美国麻省理工学院诺华-MIT连续药物生产中心从事博士后研究,合作导师为国际著名化学家、催化领域顶尖科学家、沃尔夫化学奖获得者Buchwald教授。2011年底加入西安交通大学,主要研究方向为精准催化和药物研发。已主持国家自然科学基金项目5项及陕西省杰出青年基金。至今在Chem,J. Am. Chem. Soc.,Angew. Chem. Int. Ed.,Chem. Soc. Rev.等国际著名刊物上发表SCI论文60余篇,已授权发明专利6项,参编英文专著两部。曾获亚洲核心计划讲座奖、德国蒂姆化学期刊奖、英国皇家化学会Chem. Commun.新兴科学家、陕西省“青年科技新星”、西安交通大学A类青年拔尖人才、仲英青年学者等荣誉。兼任Chinese Journal of Chemistry期刊青年编委,2019年中国化学会元素周期表年111号元素“鉨”代言人。
参考文献
- Guanghui Wang, Liang Xu, Pengfei Li*, Double N,B-Type Bidentate Boryl Ligands Enabling a Highly Active Iridium Catalyst for C–H Borylation, Am. Chem. Soc., 2015, 137, 8058–8061;
- Guanghui Wang, Li Liu, Hong Wang, You-Song Ding, Jing Zhou, Shuai Mao, Pengfei Li*, N,B-Bidentate Boryl Ligand-Supported Iridium Catalyst for Efficient Functional-Group-Directed C–H Borylation, J. Am. Chem. Soc., 2017, 139, 91–94;
- Peidong Song+, Linlin Hu+, Tao Yu, Jiao Jiao, Yangqing He, Liang Xu*, Pengfei Li*, Development of a Tunable Chiral Pyridine Ligand Unit for Enantioselective Iridium-Catalyzed C–H Borylation, ACS Catal., 2021, 11, 7339–7349;
- Shuai Zhang+, Saima Perveen+, Yizhao Ouyang+, Liang Xu, Tao Yu, Min Zhao, Linghua Wang, Peidong Song, Pengfei Li*, Design and Synthesis of Tunable Chiral 2,2’-Bipyridine Ligands: Application to the Enantioselective Nickel-Catalyzed Reductive Arylation of Aldehydes, Angew. Chem.Int. Ed., 2022, 61, e202117843.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载

















No comments yet.