
本文作者:杉杉
导读
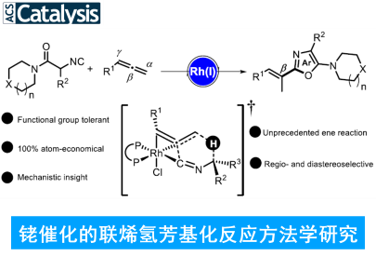
近日, 德国Albert-Ludwigs大学有机化学研究所 (Institut für Organische Chemie, Albert-Ludwigs-Universität)的B. Breit与S. Balalaie课题组在ACS Catal.中发表论文,报道首例通过铑催化剂促进的α-酸性异氰基乙酰胺与联烯之间的氢芳基化反应方法学,进而能够选择性获得相应的E-乙烯基噁唑分子。这一全新的氢芳基化策略具有优良的立体与区域选择性。同时,机理研究表明,反应过程中涉及铑催化的烯反应以及后续的还原消除步骤。
Rhodium-Catalyzed Regio- and Diastereoselective Hydroarylation of Allenes: An Unprecedented Ene Reaction
H. J. Ghazvini, H. Khosravi, S. Mirzaei, S. Balalaie, B. Breit, ACS Catal. 2021, 11, 14570. doi: 10.1021/acscatal.1c04743.
正文
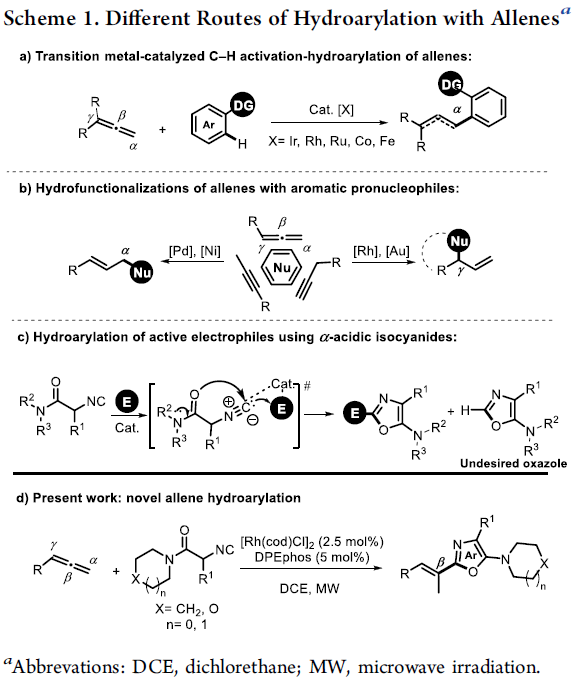
不饱和C-C键的氢芳基化反应方法学,已经成为构建取代芳基化合物的一种具有高度原子经济性的反应策略。目前,对于烯基以及炔基化合物氢芳基化方法学的研究已经有诸多的文献报道。然而,对于联烯基化合物的氢芳基化方法学却较少有相关的研究报道 (Scheme 1a-b)[1]-[6]。这里,受到通过具有α-酸性质子的官能团化异腈参与的氢芳基化反应方法学 (Scheme 1c)[7]-[8]及其在噁唑分子合成中的相关应用研究[9]-[10]的启发,B. Breit与S. Balalaie研究团队成功开发出一种铑催化的α-酸性异氰基乙酰胺与联烯之间的氢芳基化反应方法学,进而顺利实现一系列E-乙烯基噁唑分子的区域与立体选择性合成 (Scheme 1d)。
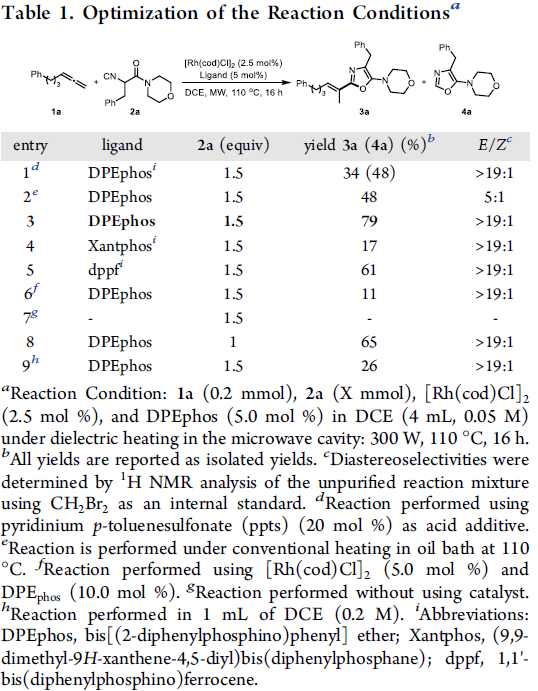
首先,作者采用1a与2a作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用[Rh(cod)Cl]2作为催化剂,DPEphos作为配体,DCM作为反应溶剂, 300 W的微波辐射,反应温度为110 oC (通过微波空腔介电加热),并获得相应的氢芳基化产物3a (79%收率,E/Z > 19:1)。
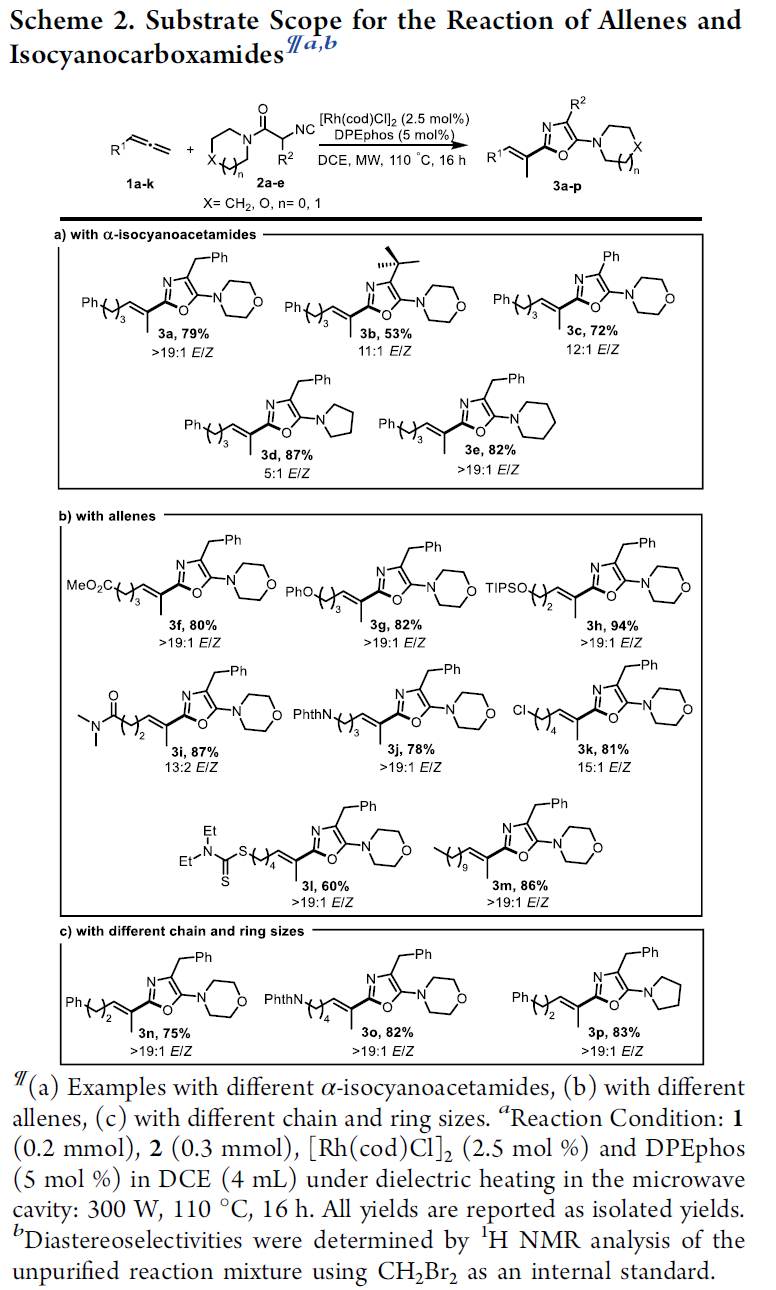
在上述的最佳反应条件下,作者首先对各类异腈底物的应用范围进行考察 (Scheme 2)。研究表明,一系列带有不同α-取代基 (例如烷基与芳基)的异腈底物,均能够顺利地参与上述的氢芳基化过程,并以高度的C2-区域选择性与中等至良好的反应收率与E-选择性,获得相应的烯基噁唑产物3a–3c (Scheme 2a)。之后,作者发现,具有不同酰胺取代基团取代的α-苄基异腈底物,同样能够有效地参与上述的氢芳基化过程,并获得预期的噁唑产物3d与3e。接下来,该小组进一步对一系列联烯底物的应用范围进行深入研究 (Scheme 2b,c)。实验表明,一系列具有不同官能团取代的联烯底物均能够与上述的标准反应条件良好地兼容,并获得相应的噁唑产物3f–3m。同时,作者观察到,联烯底物中的烷基链长对于上述氢芳基化过程的反应收率与区域选择性无显著影响,并获得相应的噁唑产物3n–3p。
接下来,为提出合理的反应机理,作者进行一系列相关控制实验与氘标记实验研究 (Scheme 3)。首先,作者发现,在上述的标准反应条件下,采用异氰基乙酸酯2h代替上述的异氰基乙酰胺底物2a时,无法顺利进行相应的氢芳基化过程。这可能源自于酰胺羰基与能够进行分子内环化步骤的酯羰基相比,具有更高的亲核性。同样地,该小组发现,采用α,α-二取代异氰基乙酰胺分子 2g时,同样无法获得预期的氢芳基化产物,进而表明异氰基乙酰胺底物中α-酸性氢的存在,对于上述氢芳基化过程的顺利进行尤为关键 (Scheme 3a)。并且,作者发现,在上述的标准反应条件下,采用噁唑衍生物4a时,上述的氢芳基化过程同样无法顺利进行。由此,能够排除在联烯底物加入之前,异腈底物首先进行关环的反应路径 (Scheme 3b)。之后,该小组进行一系列相关的氘标记实验研究。作者发现,采用DCE-d4进行的氘标记实验,在产物中并未观察到H/D交换过程的进行 (Scheme 3c)。同时,作者进一步发现,在选择α-氘代异氰基乙酰胺底物[D]-2a (90% D)参与上述的氢芳基化过程时,在相应烯基噁唑产物联烯片段中的α– (78% D)以及γ-位置(12% D)存在相应的氘标记。进而表明,通过Cα位置的金属氘化 (deuterometalation)步骤,能够直接形成最终的氢芳基化产物3a。同时,在Cγ位置通过金属氘化以及后续的烯键异构化过程,同样能够形成最终的氢芳基化产物3a (Scheme 3d)。此外,作者进一步对上述标准反应条件下, Z/E异构化过程存在的可能性进行深入研究 (Scheme 3e)。实验观察表明,在动力学控制的条件下,Z-3a无法转化为热力学更加稳定的E–3a。

基于上述研究,作者提出一种合理的反应机理(Figure 1a)。首先, [Rh(cod)Cl]2与DPEphos之间经历配体交换步骤,形成具有催化活性的[DPEphosRhCl]配合物。之后,再与联烯1以及异腈2进行配位,形成中间体A。接下来,通过中间体A的铑环丁烷化 (rhodacyclobutanation)以及后续的1,5-氢迁移过程,形成相应的铑类卡宾配合物C。再通过铑类卡宾配合物C的还原消除过程,获得相应的噁唑衍生物D,之后,D经历进一步的异构化过程,形成热力学更加稳定的产物E (在R =H, R′=alkyl时)。此外,作者进一步通过DFT计算表明,烯反应中的决速步骤为1,5-氢迁移步骤 (TS2, 总能垒为33.2 kcal/mol),进而控制产物中的氘分布以及产物中双键的相对构型 (Figure 1b)。
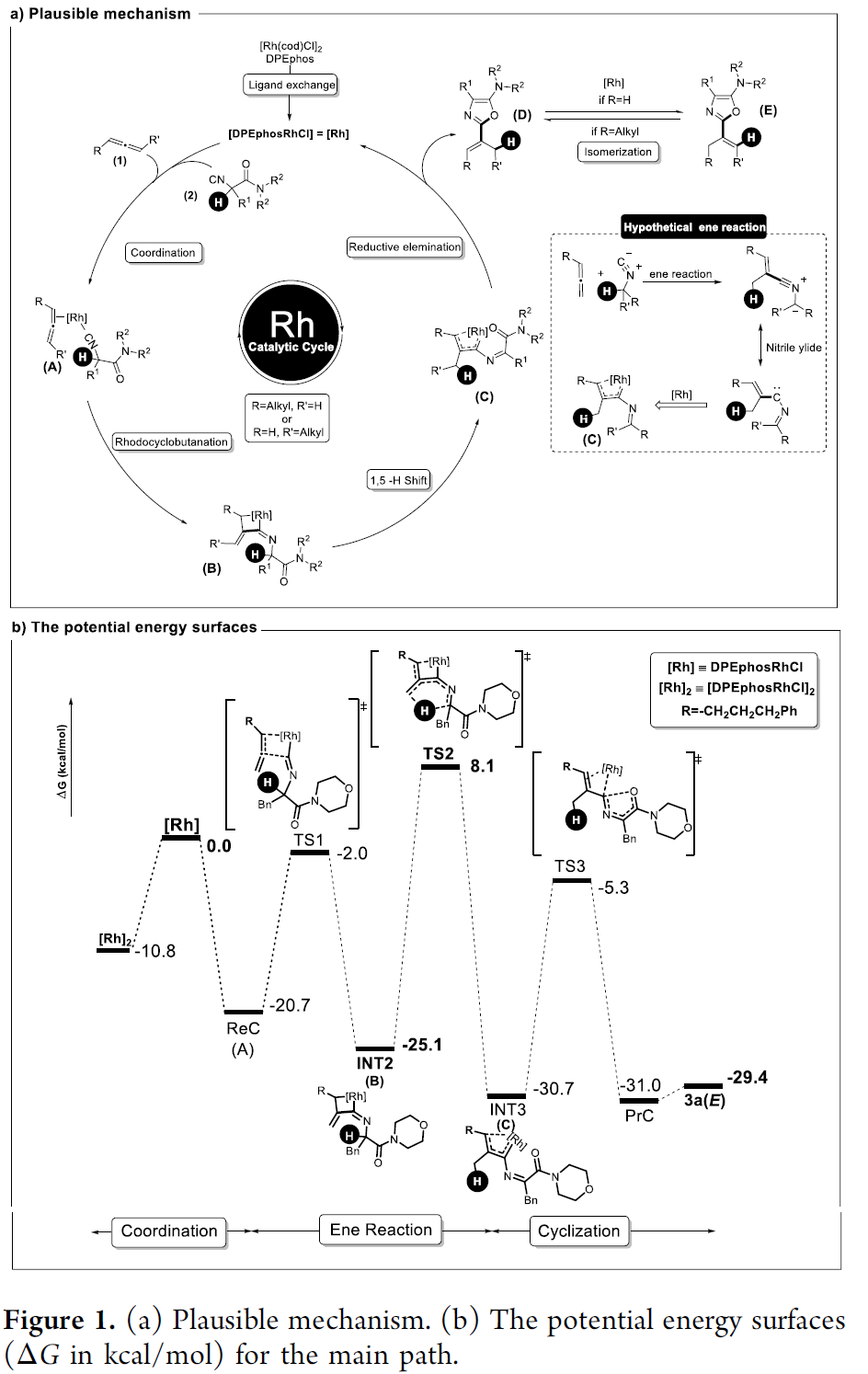
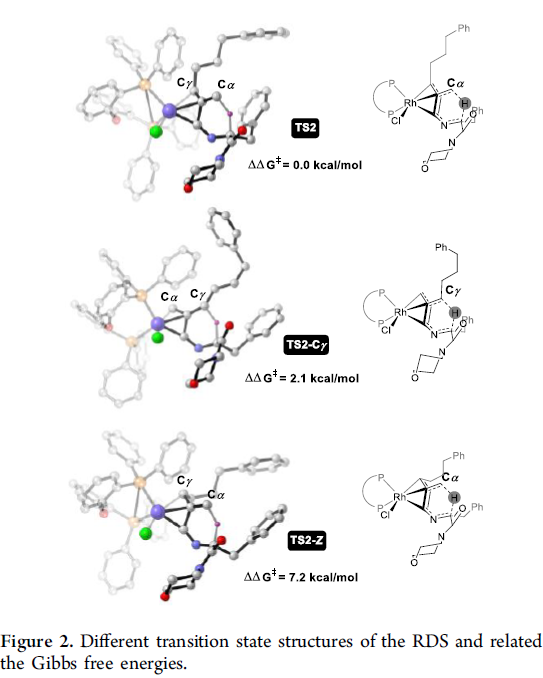
同时,该小组通过对TS2与TS2-Cγ之间的对比,进而对产物中联烯结构片段的Cα以及Cγ位置的氘分布进行深入研究。实验发现,两种过渡态之间的Gibbs自由能差为2.1 kcal/mol。同时,根据Boltzmann方程,预测Cα/Cγ氘分布的选择性为13:1 (实验值约为13:2) (Figure 2)。并且,研究表明,TS2-Z中,联烯片段的脂肪链与DPEPhos之间的立体效应以及TS与TS2之间的能量差异,决定上述氢芳基化过程的E-立体选择性。此外,作者对于端烯异构化过程,形成内烯化合物的相关计算研究,进一步证实通过上述端基异构化过程,形成热力学产物的可能性 (Figure S3)。
总结
德国Albert-Ludwigs大学的B. Breit与S. Balalaie研究团队报道一种全新的通过铑催化剂促进的各类联烯分子的氢芳基化反应方法学,进而以高度原子经济性的反应方式,成功完成一系列E-乙烯基噁唑分子的构建。同时,作者通过反应机理研究以及DFT计算,进一步证实上述的氢芳基化过程中涉及分步的烯反应以及环化过程,并表现出优良的区域选择性控制。同时,这一全新的氢芳基化策略具有良好的反应收率、高度的区域选择性以及优良的官能团兼容性等优势。
参考文献
[1] Y. J. Zhang, E. Skucas, M. J. Krische, Org. Lett. 2009, 11, 4248. doi: 10.1021/ol901759t. [2] R. Zeng, C. Fu, S. Ma, J. Am. Chem. Soc. 2012, 134, 9597. doi: 10.1021/ja303790s [3] S. Nakanowatari, L. Ackermann, Chem. – Eur. J. 2015, 21, 16246. doi: 10.1002/chem.201502785. [4] S. Nakanowatari, R. Mei, M. Feldt, L. Ackermann, ACS Catal. 2017, 7, 2511. doi: 10.1021/acscatal.7b00207. [5] A. M. Messinis, L. H. Finger, L. Hu, L. Ackermann, J. Am. Chem. Soc. 2020, 142, 13102. doi: 10.1021/jacs.0c04837. [6] D. Berthold, J. Klett, B. Breit, Chem. Sci. 2019, 10, 10048. doi: 10.1039/C9SC03894A. [7] (a) E. Novellino, G. C. Tron, J. Zhu, Chem. Soc. Rev. 2017, 46, 1295. doi: 10.1039/C6CS00444J.(b) A. D. Mathiyazhagan, G. Anilkumar, Org. Biomol. Chem. 2019, 17, 6735. doi: 10.1039/C9OB00847K.
[8] (a) A. Bhattacharyya, C. K. Shahi, S. Pradhan, M. K. Ghorai, Org. Lett. 2018, 20, 2925. doi: 10.1021/acs.orglett.8b00986.(b) X. Li, Q. Xiong, M. Guan, S. Dong, X. Liu, X. Feng, Org. Lett. 2019, 21, 6096. doi: 10.1021/acs.orglett.9b02242.
[9] (a) S. Wang, M. Wang, D. Wang, J. Zhu, Org. Lett. 2007, 9, 3615. doi: 10.1021/ol7014658.(b) R. Mossetti, T. Pirali, G. C. Tron, J. Zhu, Org. Lett. 2010, 12, 820. doi: 10.1021/ol902894p.
(c) J. Wu, W. Chen, M. Hu, H. Zou, Y. Yu, Org. Lett. 2010, 12, 616. doi: 10.1021/ol902850a.
(d) Y. Odabachian, Q. Wang, J. Zhu, Chem. – Eur. J. 2013, 19, 12229. doi: 10.1002/chem.201302106.
















No comments yet.